
Indexed In
- Academic Journals Database
- Open J Gate
- Genamics JournalSeek
- JournalTOCs
- China National Knowledge Infrastructure (CNKI)
- Scimago
- Ulrich's Periodicals Directory
- RefSeek
- Hamdard University
- EBSCO A-Z
- OCLC- WorldCat
- Publons
- MIAR
- University Grants Commission
- Geneva Foundation for Medical Education and Research
- Euro Pub
- Google Scholar
Useful Links
Share This Page
Open Access Journals
- Agri and Aquaculture
- Biochemistry
- Bioinformatics & Systems Biology
- Business & Management
- Chemistry
- Clinical Sciences
- Engineering
- Food & Nutrition
- General Science
- Genetics & Molecular Biology
- Immunology & Microbiology
- Medical Sciences
- Neuroscience & Psychology
- Nursing & Health Care
- Pharmaceutical Sciences
Review Article - (2021) Volume 12, Issue 5
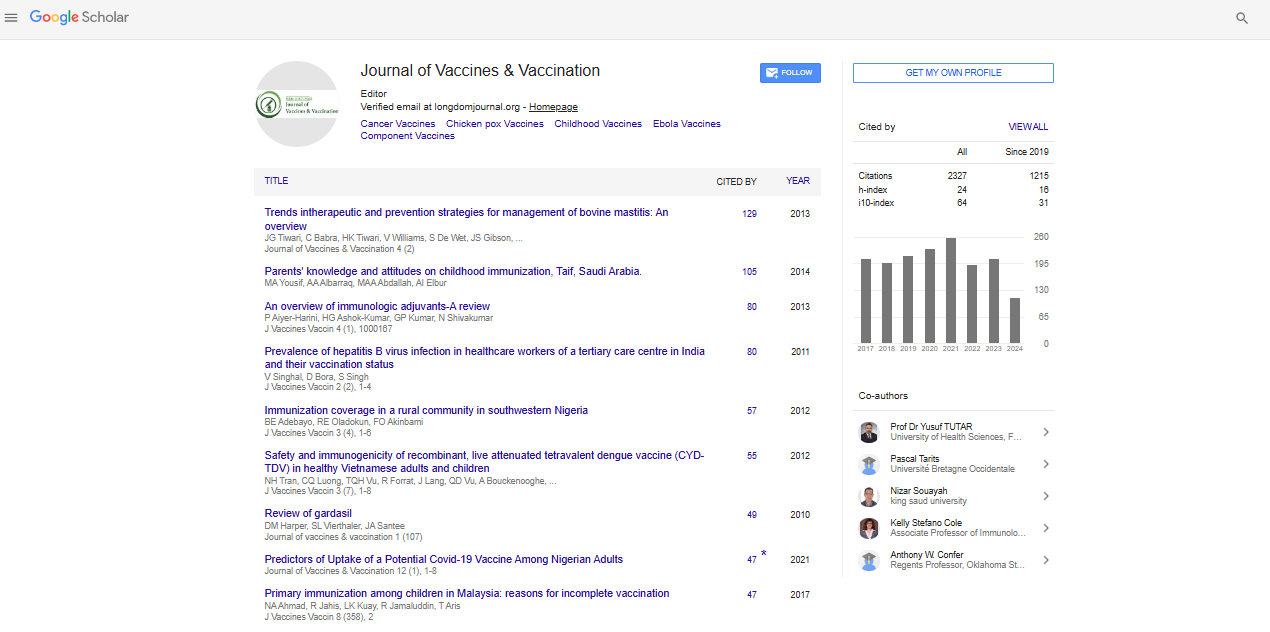
Review on Novel Bacterial Antigen Identification Methods for the Development of Candidate Vaccine
Halo Yohans*Received: 11-Aug-2021 Published: 01-Sep-2021, DOI: 10.35248/2157-7560.21.12.465
Abstract
This review mainly focused on methods that are used for identifying novel bacterial antigens that can use in recombinant subunit vaccine development. Particularly, it describes the processes involved in reverse vaccinology, genomic function, bioinformatics and immunological approaches and some associated complementary technologies such as proteomics that can be used in identification of new and potentially useful vaccine antigens. Results obtained from the application of these methods are forming basis for a new generation of vaccine for use in the control of bacterial infections of humans and animals.
Keywords
Antigen; Vaccine; Microorganism; Immunization
Introduction
Viruses and the prevention of disease and death by vaccination have profoundly improved the public health of many populations globally. Louis Pasteur, who developed the first vaccine against rabies, established in 1881 the basic paradigm for vaccine development, which included the isolation, inactivation, and injection of the causative microorganism. These basic principles have guided vaccine development during the twentieth century [1].
Improved vaccines are urgently needed to combat diseases for which current vaccination is inadequate like tuberculosis, not optimal like polysaccharide-based vaccines against pneumococcus or for which vaccination approaches have failed up to date like Meningococcus serotype B. Furthermore, there is an urging need to develop vaccines against diseases none targeted by immunization before, such as nosocomial infections caused by pathogens such as Staphylococci and Enterococci with the enormous potential to develop drug resistance. Protein based recombinant vaccine technology is considered to be the most promising approach to meet the demands of future vaccination [2].
Genomics-based strategies such as reverse vaccinology, genomic function; also bioinformatics and immonological approaches have made major contributions to vaccine development by the identification and selection of novel candidate antigens from diverse pathogenic bacteria. These approaches exploit genome sequence information in alliance with novel or custom-tailored selection technologies and display many advantages compared to conventional approaches, such as speed of identification or selection of in vivo expressed antigens [3]. Although target identification by genomics-based technologies is a real promise, the challenge lays still in the slow and laborious validation steps. As the evaluation of immune responses against candidate antigens is a crucial validation task, antigen discovery technologies that use human immunogenicity as their primary screening and selecting parameter seem to be especially valuable for vaccine development [4].
Therefore, the objective of this review is to give an account on different approaches used to identify novel microbial antigen for vaccine development and to high light their success and failure in development of vaccine against animal and human pathogens.
Literature Review
Methods for identifying antigen for the development of vaccine
Conventional method: The conventional approach to vaccine development was based on the works of Jenner and Pasteur year. Despite several successes, there are several infectious diseases for which conventional approaches for vaccine discovery have failed.
The vaccines developed for over the first two hundred diseases represents one of the most important years since Edward Jenner’s lifetime have accomplished contributions of biomedical science. With the exception striking reductions of infection and disease where ever of safe water, no other modality and not even antibiotics has applied [5].
Pasteur (1880) found that inoculation of chickens with an old culture of Pasteurella multocida protected birds against fowl cholera on subsequent challenge with fresh culture of the organism [6]. Subsequently by using biochemical, microbiological and serological, Pasteur developed an attenuated anthrax vaccine by maintaining methods [7].
Non-conventional methods: The arrival of the genome era has revolutionized subunits purified from the microorganism such as toxins vaccine development and catalyzed a shift from detoxified by chemical treatment, purified antigens or conventional culture-based approaches to genome-based polysaccharides conjugated to proteins [7]. None conventional vaccinology starts from the approaches have allowed the control of a number of whole genomic sequences and through computer analysis important infectious diseases [7] or other genomic approaches, predicts those proteins limitations [8]. The approach is widely applicable as there are many culture, [9]. Some cell-associated microorganisms require genome sequences available publicly. In the last decade, specific cell cultures for growth and this is microbial genomic sequencing has experienced an expensive extensive safety procedures for personnel exponential growth [10]. The products may be rather ill-defined and same species potentially confounding epidemiological analysis and resulting in the discovery of vaccines that may be effective under experimental conditions but ineffective in the field. These analyses illustrate how genomic sequencing is increasing our understanding of the interaction of important pathogenic microbes with their environment and facilitating the identification of relevant targets for designing vaccines that are effective under field conditions [11].
Bioinformatics: Bioinformatics is a name coined in the 1990s in response to the increase in biological information and the ease of sharing this information using the Internet. Bioinformatics is defined as “a scientific discipline that encompasses all aspects of biological information acquisition, processing, storage, distribution, analysis, and interpretation that combines the tools and techniques of mathematics, computer science, and biology with the aim of understanding the biological significance of a variety of data” [12]. Hence, bioinformatics covers a multiple of scientific disciplines and encompasses various areas of genomic analysis, including:
Computational genomics (the storage and analysis of DNA sequence using various software), proteomics (laboratory methods used to determine what proteins are actually expressed and possible cellular location), transcriptional profiling (the analysis of mRNAs at different stages of cell growth and conditions), functional genomics (combined function prediction and experimental biological analysis of gene function), structure and function determinations (predicted as well as determined 3-D structures and comparison to known protein structures and functions).
Bioinformatics, specifically computational genomics, is being applied in the search for protein subunit vaccines against bacterial diseases. Even today, with the best of medicines available, the leading cause of death in young children, worldwide, is infectious disease. Preventing infection or limiting the effects of the infection by vaccination dates back to the first scientific study and development of a safe vaccine by Jenner in 1796.
Bioinformatics use in Bacterial Vaccine discovery bio techniques cal disease in infants has recently been developed [3]. This bacterial vaccine, licensed in early 2000, provides approximately 100% protection against the seven most prevalent serotypes of S. pneumoniae. A multivalent vaccine to prevent bacterial meningitis and sepsis caused by Neisseria meningitides is also needed. In the case of this pathogen, there are at least 13 different serogroups, based on the bacterial capsular polysaccharide composition, with the majority of the disease caused by groups A, B, C, Y, and W-135 [13,14].
Polysaccharide based vaccines are available (for older children and adults) against serogroups A, C, Y, and W-135, and a serogroup C conjugate vaccine is available in several European countries. Unfortunately, the serogroup B capsular polysaccharide is immunobiologically similar to polysialic acid expressed on human cells and is poorly immunogenic in humans. Thus, trying to generate antibodies to this polysaccharide could potentially induce autoimmune disease and harm the vaccine. Therefore, a protein antigen based vaccine is being pursued [15].
For organisms that make no capsular polysaccharide or do not produce toxin-mediated diseases, other targets have to be identified. The most promising candidates are surface-exposed or secreted proteins. In the “pre-genomic sequencing era”, a common approach used in early vaccine discovery programs was to generate polyclonal or monoclonal antisera in animals by administering either live or inactivated bacteria. The sera were analyzed in an immuno-assay format against either whole bacteria or a bacterial protein preparation. The major reactive proteins, usually less than a dozen, were further characterized by purifying the native proteins and analyzing the subsequent sera, generated from vaccinating animals with the same proteins. The sera were tested in functional immunobiologically assays and then in an animal model, the proteins that elicited functional immunobiological activity [16].
Bioinformatics and computational vaccinology designing an ideal vaccine depends greatly on several factors associated with targeted pathogens and host responses, including knowledge at the molecular level of the immune response, pathogenesis, hostpathogen interaction, and genetic and physiological variation among animals and pathogens. Recently discovered genome sequences of food animals and pathogens together with rapid advances in biotechnology will allow us to collect an unprecedented amount of information on hosts and pathogens that may have significant implications [17].
Define predicted coding sequences Amino acid similarity searching (BLAST, etc.) Similar to human proteins Pseudogenes Putative virulence determinants Anomalous G+C regions Secreted, lipoproteins, outer membrane, periplasmic Clone and express Expressed proteins Immunological and biological assays Vaccine candidates! Cytoplasmic Localization prediction (PSORT, signal P) Hypothetical or unknown function. The initial step involves annotating the genome to define its coding capacity and hence all potential antigens. Bioinformatics similarity searches should then be performed to discard pseudogenes and anything resembling human proteins. The remaining list of genes is then cloned and expressed and analysed immunologically for vaccine candidate’s vaccine discovery. However, transforming this information into practical understanding requires intensive datamining using sophisticated computational and bioinformatic tools. Highly intensive computation using high-speed central processing unit, multithread, and 64 bit technologies have greatly facilitated this process and using computational approaches in vaccine design has become known as ‘computational vaccinology’ [17]. Applying bioinformatics algorithms to facilitate vaccine design is a very powerful approach that is changing many paradigms of vaccine discovery [18].
Genomics: The availability of a complete microbial genome sequence in 1995 marked the beginning of genomic era that has revolutionized vaccine development and catalyzed a shift from conventional culture-based approaches to genome-based vaccinology. Therefore, potentially surface-exposed proteins can be identified in a reverse manner, starting from the genome rather than from the microorganism [19].
Genomics also has the capability to make DNA vaccination studies much more efficient. Before the genome sequence was available for Mycobacterium avium subspecies Para tuberculosis DNA vaccination was attempted for cattle pathogen using the expression library immunisation procedure [20]. This study revealed two pools of DNA that were shown to be protective in mice and limited efforts were made to identify the relevant DNA in those pools [21].
Random expression library immunisation was used because the genome sequence was not available at the project’s inception. This random cloning method meant that the majority of clones would be in the opposite orientation relative to the coding strand or would be out of frame with the coding sequence. Therefore, many additional clones were needed to make the library truly representative of every coding sequence in the genome and an approximate total of 16,500 clones were used to immunize mice in that study [21].
With the genome sequence now complete [22], a directed expression library immunisation project, in which each clone faithfully represented a single coding sequence, could be initiated. This method has the advantage that fewer clones are needed, making resulting clone pools less complex and there is no ‘junk’ or non-functional clones such as those in the opposite orientation or out of frame. For such a study, only 4,350 clones would be needed because that is the total number of genes present in the M. avium subspecies paratuberculosis genome.
An added benefit is that fewer mice would be needed to test the clone pools [22].
Genomic approaches can also identify the best targets for knockout mutations that enable engineering of attenuated vaccine strains. However, as yet, there are no published studies for bacterial pathogens that demonstrate a genome wide approach that can identify a target, knockout and show both attenuation and protection in an animal host. Rather, the literature reports studies in which genomics has been used to genetically define a known vaccine strain [23].
The most famous example of the use of genomics to define an attenuated bacterial vaccine strain is M. bovis BCG (Bacillus Calmette-Guerin-named after the French scientists Calmette and Guerin), which is the most widely used global vaccine to prevent human Tuberculosis (TB). Over 3 billion individuals have been vaccinated with BCG without major side effects [24]. The BCG vaccine strain was derived long ago from a fully virulent isolate of M. bovis by prolonged serial passage of the bacterium resulting in its attenuation [25]. However, the molecular basis for this attenuation was never understood until the complete genome sequences of M. tuberculosis , the causative agent of TB, and M. bovis BCG became available [26]. Genomic comparison of these two species revealed one region of deletion in BCG, termed as RD1. This region contains the well-known antigen ESAT-6, a secreted antigenic target that strongly induces Th1 immune responses. More than any other benefit, whole genome analysis of pathogens enables the targeted selection of protective immunogens encoded by the disease-causing pathogen. This allows investigators to move away from empirical approaches in vaccine development towards a more focused, logical development and discovery of protective DNA segments and proteins [27].
Reverse vaccinology: With the arrival of whole-genome sequencing, genome-based antigen selection has played a major role in antigen discovery and vaccine design. One approach that has been used to mine pathogenic bacterial genomes has been coined “Reverse vaccinology,” and allows the investigation of the complete potential antigenic repertoire of an organism from its genome sequence. Reverse vaccinology involves the cloning and expression of all the proteins in an organism’s genome sequence that are predicted in silico to be surface exposed or secreted. Then each protein is screened, through high-through put immunization, for their ability to elicit antibodies in mice that can kill or neutralize the target organism. The first pathogen addressed using reverse vaccinology was N. Meningitides serogroup B (MenB) [28], for which no broadly protective vaccine exists owing to the similarity of its capsular polysaccharide to a self-antigen and the hyper variability of its major outer membrane protein antigens. This approach led to the identification of 29 novel antigens that can elicit bactericidal antibodies against the pathogen in vitro . After successful preclinical studies, the vaccine deriving from the genome approach entered phase I testing in 2002 [29].
In November 2012, the European Medicinal Agency recommended the granting of a marketing authorization for Bexsero, the first vaccine to provide broad coverage against meningococcal serogroup B. This was followed by the approval of the European Commission in January 2013. The three main antigens identified by reverse vaccinology that are formulated as components of the MenB vaccine, Neisserial Heparin-Binding Antigen (NHBA), factor H-binding protein (fHbp), and N. meningitidis adhesin A (NadA), have all subsequently been implicated as playing important roles in meningococcal virulence [30-33]. Now we use a genome-based approach in reverse vaccinology where the genome sequence of a pathogen is screened with bioinformatic tools to identify open reading frames that may encode candidate proteins. Proteins predicted to be surface-exposed or secreted are considered as vaccine candidates for further laboratory testing. Some proteins having structures similar to known toxins can also be included in the candidate list. If the genome sequences of different strains (virulent and avirulent) or serotypes are available, a pan-genome approach can also be used to identify candidate vaccines by comparative genomics. The applications of these approaches in vaccine development have been reported [34].
Since the first application of reverse vaccinology, based on the single genome of a MenB strain [28], various advancements have been made to this genomic approach. Amultigenome or pangenome reverse vaccinology approach was applied to Group B Streptococcus (GBS) to identify antigens from the extended gene repertoire of the species rather than from a single organism [35], and the subtractive reverse vaccinology approach has been used to identify antigens present in pathogenic but not commensal strains of Escherichia coli [36]. Reverse vaccinology has been applied to a wide range of bacterial pathogens and has provided a long list of promising antigens from functionally blind interrogation of their genomes, and the subsequent studies on antigen function are leading to increased understanding of the biology of the pathogens [37].
Recently, subtractive reverse vaccinology has been applied to E. coli and five out of the nine protective antigens identified are located on putative pathogenicity islands specific for pathogenic E. coli strains. For the rapid identification of vaccine targets the reverse vaccinology is generating promising results for various microorganisms. As the genomic era progressed, reverse vaccinology has evolved with a pan-genome approach and the advantage of multi-strain genome analysis was highlighted by the discovery of universal vaccine candidates against disease-causing isolates of Streptococcus agalactiae. Moreover, for those bacterial species that exist in both as commensal and pathogenic forms, by comparing the genome sequences of the two types, one can identify pathogenic specific traits. In this regard, subtractive reverse vaccinology has been applied to identify antigens produced only in pathogenic strains of Escherichia coli species. In general, currently observed excellent results of reverse vaccinology can be good indicators of the potential of genome based approach for vaccine development. Thus, the ever-growing body of genomic data and new genome-based approaches should play a critical role in the future to enable timely development of vaccines against infectious diseases [37].
The reverse vaccinology approach has been applied to other pathogenic bacteria including Bacillus anthracis, Porphyromonas gingivalis, S. pneumoniae, Chlamydia pneumoniae, and Brucella melitensis . All the results obtained so far showed that genome mining allowed to increase the number of candidate vaccine antigens by several orders of magnitude. However, during the development of new vaccine antigens through reverse vaccinology, we understood that parallel genomebased studies can be applied in order to better characterize the candidates and understand their potentials. In the development of a universal vaccine capable of inducing protection against virtually all circulating strains, accurate characterization of the selected vaccine candidates is highly recommended. The analysis of the sequence conservation of a given antigen is a fundamental aspect in its evaluation as vaccine candid [38].
Functional genomics approach: Functional genomics linking genotype, through transcriptomics and proteomics, to phenotype has been applied to many pathogens to identify genes essential to survival or virulence that may be valid vaccine candidates. Functional genomics is the study of the functional expression of an organism’s genomic information [39]. The aim of functional genomics is to reveal the links between a specific genotype and its corresponding phenotype. The phenotype results from the expression of genes through conversion into systemic, catalytic and regulatory products and is a complex function of genotype and environment [40].
During this approach different techniques are used. These techniques are:
Signature Tagged Mutagenesis (STM): STM developed by David Holden, is an approach based on random mutagenesis to identify genes required for in vivo survival [41], and therefore, a powerful tool for identifying potential virulence factors. Selection technique uses a collection of transposons, each one modified by the incorporation of a different DNA sequence tag of approximately 40 bp. In this way, each mutant can be recognized by the specific sequence present in the integrated transposon. Tagged mutants strains are pooled together and inoculated into an animal. After infection is established, bacteria are recovered from the infected animals and plated. Mutants that are attenuated will not be recovered from the animals. Comparison of tags that are present in the inoculum but absent from the recovered bacteria identifies attenuated mutants. There are two advantages of this approach for the design of novel vaccines. On the one hand, the technique potentially allows the identification of attenuated mutants that fail to cause productive infection and hence may be used as live vaccines. On the other, proteins identified as being essential for infection or disease is likely to be good candidates for subunit vaccines. STM has been utilized to discover virulence genes from a large variety of bacterial species including Mycobacterium tuberculosis, Staphylococcus aureus, Salmonella typhimurium, Vibrio cholerae, Yersina enterocolitica [42-45], and more recently Streptococcus agalactiae [46] and N. meningitidis [45].
In vivo Expression Technology (IVET): The IVET strategy requires a bacterial strain carrying a mutation in a biosynthetic gene that attenuates growth in vivo , for example, a purA auxotroph. The biosynthetic function, essential for growth in the host, is provided by a promoterless purA gene, in which fragments obtained from a random library of the pathogen’s chromosomal DNA supply the missing transcription elements.
The positively selected fusions are then sequenced to identify in vivo induced genes. This IVET method necessities the existence of an attenuating and complement able auxotrophy, which may not be available in all microbial systems. However, alternative systems have been proposed based on reporter genes encoding resistance to antibiotics [47], or encoding the Green Fluorescent Protein (GFP). This latter method is known as Differential Fluorescence Induction (DFI) [48,49].
IVET technology has been successfully utilized to identify several virulence genes in different human pathogen such as Pseudomonas aeruginosa, S. typhimurium, Y. enterocolitica, V. cholerae and S. aureus [50,51]. It is important to point out that both IVET and STM technologies have been applied before the advent of the genomic era. In fact, the previous knowledge of the genome sequence is not strictly necessary for their application. Once the mutants are selected, the regions flanking the insertions are sequenced and the inactivated gene is identified. However, both technologies greatly benefit from the availability of genome sequences since the identification of the inactivated genes can be carried out rapidly by sequencing only few nucleotides upstream and downstream from the insertions.
Advent of metagenomics: Its application to the study of the human microbiome now provides a unique opportunity to perform these comparisons, as the total gene pool from whole microbial populations can be compared against the genome of individual pathogenic strains. A fast way to make these comparisons is achieved by metagenomic recruitments [52]. Individual metagenomic reads that give a hit over a certain identity threshold against a reference bacterial genome are ‘‘recruited’’ to plot a graph which will vary in density depending on the abundance of that organism in the sample. Interestingly, it has frequently been found that recruitments of marine bacteria against all marine metagenomes available identified several ‘‘islands’’ of extremely limited or absent coverage, even for species which were dominant in the sample [53]. These ‘‘Metagenomic Islands’’ have also been found in other free-living environments and represent segments of the genome which are highly variable or specific to the reference strain. Assuming that virulent strains are absent from healthy individuals, metagenomic recruitments of pathogenic strains of bacteria whose commensal counterparts are typically found in the human microbiome should reveal MIs at the regions where virulence genes are located. Results Human-associated bacteria display Metagenomic Islands (MI) similarly to free-living bacteria, when metagenomic recruitments are made between the genomes of gut-associated bacteria against the human gut metagenome, regions with low or absent recruitment are clearly visible. This shows that gut inhabitants also have genomic regions that appear to be unique to individual strains [54].
However, the appearance of MIs could also be showing genes specific to virulent strains because recruitment plots for commensal bacteria displayed a higher coverage along the genome and a limited presence of MIs, which were limited to mobile genetic elements, mainly phage genes (48.8% of the total) and several outer membrane proteins (14.6% of the total). Thus, in order to determine whether regions of absent recruitment identify genes involved in pathogenicity, we performed a systematic description of gene content in MIs from human pathogens for which pathogenicity islands and virulence genes are well characterized. The gene content of all MIs identified in pathogenic Shigella and Escherichia strains against the gut metagenome also reveals, as expected, the presence of mobile elements like IS elements and phage genes. In fact, prophages appear to be quite unique to individual strains and represent an important portion of Mis [54].
Combination of proteomics: This has been widely applied to develop two-dimensional polyacrylamide gel electrophoresis maps and databases, evaluate gene expression profiles under different environmental conditions, assess global changes associated with specific mutations, and define drug targets of bacterial pathogens [55]. When coupled to immunological assays, proteomics may also be used to identify B-cell and T-cell antigens within complex protein mixtures. As shown in the present study, the combination of proteomics and Western- Blotting (WB), i.e., immune proteomics, helped us identify dominant immunogens from membrane proteins that hold promise as vaccine candidates. This rapid and efficient method for identifying vaccine candidates differs from other reports in that vaccine candidates were determined by practical evaluation of immune reactivity rather than by theoretical analysis of genes and proteins based on bioinformatics [56,57].
While the availability of the complete genome sequence permits the identification of all potential protein products, this information is not sufficient to allow the identification of the subset of proteins (the proteome), which are actually expressed at any stage of the life of the bacteria in particular compartments or under different growth conditions.
Recently, advances in protein separation technologies, combined with mass spectrometry and genome sequencing, have made the elucidation of total protein components of a given cellular population a feasible task [58]. Moreover, the combination of proteomics with serological analysis has recently led to the development of a new valuable approach defined as Serological Proteome Analysis (SERPA) for the identification of in vivo immunogens suitable as vaccine candidates [59].
An attractive and powerful application of proteomics was recently described by Grandi and colleagues who analyzed the surface proteome of Streptococcus pyogenes (Group A Streptococcus, GAS) to identify new vaccine candidate proteins.
This new approach, consisting of the surface digestion of live bacteria with different proteases, allowed fast and consistent identification of proteins that are expressed on the bacteria surface and thus exposed to the immune system. The cell-surface peptide fragments generated after protease treatment of GAS strain SF370 were recovered, concentrated and analyzed by tandem mass spectrometry and identified using bioinformatic examination of the publicly available genome sequence. Seventytwo proteins were identified, of which only four were predicted by the PSORT algorithm to be cytoplasmic proteins indicating that the method was highly specific for surface-exposed proteins [59].
Proteomics in vaccine design with the availability of genomic sequences, the progress achieved in 2D-gel electrophoresis separation techniques and advances in mass spectrometry analysis means that it is now possible to separate, identify and catalogue the proteins expressed in a cell under several conditions. The entire set of proteins encoded by the genome has been defined as “proteome” [60]. In proteome analysis, a protein mixture (e.g. outer membrane preparations or whole cell lysates) is first resolved in its individual components using separation procedures. Once separated, each protein undergoes digestion with a specific protease to generate discrete peptide fragments of which the molecular masses can be accurately evaluated by mass spectroscopy. The experimental result is then compared with theoretical results expected for the same specific degradation of all predicted proteins from the genome sequence. In this way, the protein can be unequivocally identified as the product of a specific gene. Physical analysis of the proteome permits the identification of proteins actually expressed in a particular compartment or under different conditions of growth. Recently, this approach has been used to identify novel bacterial vaccine candidates against several human pathogens [56].
Proteome analysis of the outer surface proteins of the human pathogen S. agalactiae allowed the discovery of novel surface proteins. Sera, raised against some of these proteins, were protective in a neonatal-animal model system against a lethal dose of bacteria. Hence novel potential vaccine candidates against S. agalactiae were identified [61]. Grandi and colleagues have recently combined genome mining and proteome technologies to identify surface-exposed antigens of Chlamydia pneumoniae [62]. The authors identified 157 putative surfaceexposed proteins by in silico analysis of the pathogen genome. They then used recombinant forms of these proteins expressed in E. coli to raise antisera that were used to assess surface location by flow cytometry. Finally, 2D gel electrophoresis and mass spectroscopy were used to confirm the expression of the antigens in the elementary body phase of development. The result of this systematic genome–proteome combined approach represents the first successful effort to define surface protein organization of C. pneumoniae and opens the way to the selection of suitable components for a novel vaccine. Finally, proteome comparisons are likely to become progressively more important for the study of bacterial pathogenesis; comparison of virulent strains of a pathogen with non-virulent or commensal strains of the same bacteria should permit the identification of proteins involved in virulence Jungblut et al.
DNA microarray: Microarrays that provide hybridisation targets representing the entire genome, all placed on a microscope slide. Microarrays allow investigators to assess genetic variation between isolates and characterise global patterns of gene expression. For microarray analysis, RNA or DNA samples are differentially tagged with chemical labels and used to hybridise with DNA targets on the array. Unhybridised material is removed by washing and the retained, tagged samples are modified with a chemical that fluoresces when excited by lasers in a specialised instrument. The intensity of each spot, representing 26 hybridisation target, usually a specific gene, is measured and compared to control samples to determine either genetic diversity (DNA input) or differential gene expression (RNA input). For example, genetic differences among M. avium subspecies paratuberculosis were identified by microarray studies [63], resulting in valuable information on bacterial adaptation to different mammalian hosts. Microarray analysis of Pasteurella multocida gene expression during growth in chickens revealed a subset of genes induced by infection that are also expressed in response to iron limitation [64]. Similar studies using in vitro models have also helped characterise changes in gene expression (e.g. temperature response in L. interrogans) [65] and invasion of bovine epithelial cells by M. avium subsp. paratuberculosis [63] and are helping to expand our understanding of how bacteria respond and adapt to growth in the natural host. A key point in using microarrays to study gene expression is that many putative genes identified by genomic analysis encode proteins of unknown function. By identifying genes that respond to environmental stimuli rather than selecting genes based on a bias formed by presumed function, it may be possible to identify bacterial proteins essential for survival in the host. This information is critical for rational selection of proteins for development as subunit vaccines [63].
Microarray analysis has been used to genotype bacteria, viruses and parasites via Comparative Genome Hybridization (CGH). CGH involves the use of a microarray containing DNA from a sequenced reference microorganism. These arrays can be used to compare genomes of different unsequenced isolates by detecting genes that are conserved between them. However, this highlights one intrinsic technical limitation of microarray: detection is limited to the DNA spotted on the array. This method also fails to detect acquisition events with respect to the reference strains. Alternatively, microarrays can be used to study gene expression. In this case they are hybridized with cDNA prepared from mRNA isolated from microorganism grown under different growth conditions (for example in vivo versus in vitro growth). Researchers are using microarray technology to identify genes that are differently expressed in response to alteration in environmental parameters and to evaluate mutations or key factors in regulatory and metabolic pathways. Another purpose is to capture the transcriptome of bacteria growing within infected cells, tissues or animal models [66,67]. Gene expression can be analyzed in either pathogen or host, thus allowing investigation of both sides of the host–pathogen interaction [68-70].
The first example where microarray technology was successfully used to identify potential vaccine candidates, as well as new virulence genes, was in the case of N. meningitidis, where DNA microarray technology was used to study gene regulation after interaction of N. meningitidis to human epithelial cells [71]. RNA was isolated from adherent and non-adherent bacteria and comparatively analyzed on DNA microarrays carrying the entire collection of PCR-amplified meningococcus B genes.
DNA microarray technology appears to be particularly attractive for the analysis of organisms with relatively simple genomes, as in the case of bacteria. DNA chips carrying the entire bacterial genome can be easily prepared, allowing whole genome expression analysis. Bacterial microarrays promise to be particularly helpful in new virulence gene hunting. The complex interaction between host and pathogen is now being explored using microarrays [72,73]. Virulence gene expression can be monitored by growing the pathogens in the appropriate in vivo models (cell cultures and/or animals) and, after recovering the bacteria for RNA preparation and labelling, the gene activity is analysed and compared with the expression of the genes under in vitro conditions. By following the pattern of gene expression at different times, it is possible to elucidate all of the host genes and those of the bacteria whose expression is modified (up or down regulated) during host-pathogen interaction [74].
In an independent work analysed the transcriptional changes in N. meningitidis in a model system of three key steps of meningococcal infection. RNA was isolated from meningococci incubated in human serum as well as adherent to human epithelial and endothelial cells. With this approach, authors found a wide range of surface proteins, which are induced under in vivo conditions. These antigens could represent novel candidates for a protein-based vaccine for the prevention of meningococcal disease [75]. The whole genome view of microarrays makes them promising tools to identify candidate genes as targets for vaccine or drug development. However, because the results of pathogen gene expression and of hostpathogen interactions are influenced by the model system used, such results must be interpreted cautiously. In addition, expression data have limitations because mRNA levels may not reflect protein levels, and expression of a protein may not always have a pathological consequence [76].
Consequently, traditional biological, pathology and toxicity studies remain necessary. DNA microarrays containing the genome of one strain can be hybridised with total genomic DNA from different strains or related bacteria for which genome sequence data do not exist thus permitting the identification of genes present in one strain and absent in another [77]. This technique to compare the genomes of 22 strains of S. agalactiae (Group B streptococcus), comprising examples of all nine known serotypes, with the genome of a serotype V isolate of which they had determined the complete genome sequence. The analysis revealed a number of regions of the genome that are highly variable and, more importantly, those genes common to all strains. This latter group contains the best candidates for a vaccine capable to induce cross serotype protection [78].
Immunomics-based: Immunomics is the study of the set of antigens, especially T and B-cell epitopes, which are recognized by host immune systems including human or animal hosts. Many immune informatics algorithms have been invented to predict T and B-cell immune epitopes. T-cell epitopes are bound in a linear form to MHC class I or class II molecules. T-cell epitopes can be predicted with high accuracy [79].
B-cell epitopes can be linear or nonlinear (or called conformational). It remains a huge challenge to computationally predict B-cell immune epitopes. Currently, the best accuracy of predicting linear B-cell epitopes is approximately 60%-70%. Over 90% of B-cell epitopes are nonlinear and require the knowledge of native 3D protein structure. There has not been proper approach achieving high performance in nonlinear B-cell epitope prediction. It is noted that computational epitope prediction-based Immunomics methods have often been integrated with other omics technologies. For example, using DNA microarray data developed a matrix-based computational algorithm to successfully predict a list of immunogenic epitope peptides uniquely associated with colon cancer. These peptides are likely vaccine targets for development of a colon cancer vaccine Sturniolo et al. If candidate antigens are identified, peptide vaccines can be developed based on the epitopes of the antigens. ‘Immunoinformatics’ the new science of epitope prediction applies bioinformatics to the design of peptide vaccines [80].
Antigen processing and presentation in the adaptive immune response are well-known at the molecular level. B-cell epitopes can be either linear or discontinuous amino acid residues dependent on the conformation of protein antigens (surface accessibility), whereas T-cell epitopes are short linear peptides that are processed by proteases and presented by class I and II Major Histocompatibility Complex (MHC) molecules. These epitopes can be mapped using laboratory procedures, which are costly and labour intensive. The epitopes can also be predicted using various bioinformatic algorithms. Currently, T-cell epitopes are more predictable than B-cell epitopes due to the linear nature of the former. The prediction of T-cell epitopes can be based on anchor motifs in the binding pockets of MHC molecules [81], or on training sets of laboratory tested data, using statistical methods such as a hidden Markov model or machine learning methods, e.g. artificial neural networks and support vector machines [82]. A protein called ‘transporter associated with antigen presentation’ selectively transports endogenous antigenic peptides into the Endoplasmic Reticulum (ER) for class I MHC antigen presentation. This selectivity can be taken into consideration in the prediction of class I MHC epitopes [83]. In contrast to T-cell epitopes, B-cell epitopes remain much less predictable [84].
Recently, using recurrent neural network [85], machine learning classifiers [86] and structural-energetic analysis [87] improved the prediction of continuous B-cell epitopes, whereas the combination of protein 3D structures and statistics has been used to predict discontinuous B-cell epitopes [88]. Although the technical difficulties of predicting B-cell epitopes remain to be overcome, combining laboratory and bioinformatic analysis, such as phage display and mimotope analyses, can increase the accuracy of predicting continuous and linear epitopes mimotopes were first described as peptides that mimic native epitopes of foot and mouth disease virus and can bind to the same antibody as native antigens. Candidate vaccines can be identified based on mimotopes that can induce antibody capable of binding to native antigens of pathogens [89]. This approach may be useful for developing multi-epitope vaccines to fight against pathogens with several serotypes, such as foot and mouth disease virus. One of the challenges of epitope-based vaccines is population coverage due to MHC polymorphism. Different MHC molecules display distinct peptide-binding specificity [90]. However, it has been shown that certain MHC alleles share overlapping peptide-binding specificity and the alleles can be grouped into super types based on their common binding specificity. Predicting peptides that bind to MHC super types for vaccine development can avoid the complication of MHC polymorphism. MHC alleles can also be grouped into supertypes based on the bioinformatics analysis of MHC protein structures and sequences [91], and supertypic MHC ligands can be predicted for multi-epitope vaccine development to increase population coverage [92]. It has been estimated that targeting only 3 to 6 class I HLA alleles should cover ~90% of the human population because of linkage disequilibrium in the MHC loci. MHC genes are also tightly linked in food animals another application of bioinformatics in vaccine development is the interpretation of data collected with functional genomics approaches to gain detailed understanding of the immune response, pathogenesis, and host–pathogen interaction [93-97].
Discussion and Conclusion
With the growing problem of antimicrobial resistance, and newly emerging or reemerging pathogens we are increasingly looking to the development and use of vaccines to control infectious diseases. Furthermore, new and improved vaccines are needed to replace several vaccines that are suboptimal in terms of efficacy or safety. Advancing technologies continue to transform the field of vaccinology, and we are now able to use genomic-based approaches to aid selection of vaccine candidates and structure-based design to optimize the chosen immunogens. In parallel, the increasing use of systems biology will provide essential insights into the immune response elicited by vaccines, and should help identify signatures of immunogenicity and correlates of protection. This improved understanding of both the host and the pathogen is aiding the development of new vaccine technologies, including the use of small molecule adjuvants to target specific immune responses, as well as new delivery systems and immunization schemes to optimize vaccine efficacy, which are essential components for the next generation of vaccine development. The knowledge gained through systems biology will also increase fundamental understanding of microbial pathogenesis enabling continued advancements in vaccine development.
REFERENCES
- Telford JL, Barocchi MA, Margarit I, Rappuoli R, Grandi G. Pili in gram-positive pathogens. Nat Rev Microbiol. 2006;4(7):509-519.
- Kurz S, Hübner C, Aepinus C, Theiss S, Guckenberger M, Panzner U, et al. Transcriptome-based antigen identification for Neisseria meningitidis. Vaccine. 2003;21(7-8):768-775.
- Serruto D, Rappuoli R. Post-genomic vaccine development. FEBS letters. 2006 ;580(12):2985-2992.
- Medini D, Serruto D, Parkhill J, Relman DA, Donati C, Moxon R, et al. Microbiology in the post-genomic era. Nat Rev Microbiol. 2008;6(6):419-430.
- Plotkin SA. Vaccines: the fourth century. Clin Vaccine Immunol. 2009 ;16(12):1709-1719.
- Tomar D, Chattree V, Tripathi V, Khan AA, Bakshi AR, Rao DN. New dimensions in vaccinology: A new insight. Indian J Clin Biochem. 2005;20(1):213-230.
- Serruto D, Spadafina T, Ciucchi L, Lewis LA, Ram S, Tontini M, et al. Neisseria meningitidis GNA2132, a heparin-binding protein that induces protective immunity in humans. Proc. Natl. Acady. 2010;107(8):3770-3775.
- World Health Organization (WHO), State of the world’s vaccines and immunization. 3rd Edition, Geneva, Switzerland. 2009; 20-25.
- Morgan AJ, Parker S. Translational mini review series on vaccines: the Edward Jenner Museum and the history of vaccination. Clin Exp Immunol. 2007;147(3):389-394.
- Pasic L, Rodriguez-Mueller B, Martin-Cuadrado A, Mira A, Rohwer F, Rodriguez Valera. Metagenomic islands of hyperhalophiles: the case of Salinibacter ruber. BMC Genomics.2009; 10(1): 570.
- National Center for Biotechnology Information (NCBI) Microbial Genomes, 2010.
- Benton D. Bioinformatics-principles and potential of a new multidisciplinary tool. Trends Biotechnol. 1996;14(8):261-272.
- Black S, Shinefield H, Fireman B, Lewis E, Ray P, Hansen JR, et al. Efficacy, safety and immunogenicity of heptavalent pneumococcal conjugate vaccine in children. Pediatr Infect Dis J. 2000;19(3):187-195.
- Hoiseth S. (2000).Encyclopedia of Microbiology, In J. Lederberg. 2000;2(4):767-778.
- Rappuoli R. Pushing the limits of cellular microbiology: Microarrays to study bacteria–host cell intimate contacts. Proc. Natl. Acad. 2000 Dec 5;97(25):13467-13469.
- Rappuoli R. Conjugates and reverse vaccinology to eliminate bacterial meningitis. Vaccine. 2001;19(19):2319-2322.
- Flower DR. Databases and data mining for computational vaccinology. Curr Opin Drug Discov Devel. 2003;6(3):396-400.
- Serruto D, Rappuoli R. Post-genomic vaccine development. FEBS letters. 2006 ;580(12):2985-2992.
- Fleischmann RD, Adams MD, White O, Clayton RA, Kirkness EF, Kerlavage AR, et al. Whole-genome random sequencing and assembly of Haemophilus influenzae Rd. Science. 1995;269(5223):496-512.
- Johnston SA, Barry MA. Genetic to genomic vaccination. Vaccine. 1997;15(8):808-809.
- Huntley JF, Stabel JR, Paustian ML, Reinhardt TA, Bannantine JP. Expression library immunization confers protection against Mycobacterium avium subsp. paratuberculosis infection. Infect Immun. 2005;73(10):6877-6884
- Li L, Bannantine JP, Zhang Q, Amonsin A, May BJ, Alt D, et al. The complete genome sequence of Mycobacterium avium subspecies paratuberculosis. Proc Natl Acad Sci U S A. 2005;102(35):12344-12349.
- Min W, Lillehoj HS, Ashwell CM, Van Tassell CP, Dalloul RA, Matukumalli LK, et al. Expressed sequence tag analysis of Eimeria-stimulated intestinal intraepithelial lymphocytes in chickens. Mol biotechnol. 2005;30(2):143-149.
- Bloom BR, Murray CJ. Tuberculosis: Commentary on a reemergent killer. Science. 1992;257(5073):1055-1064.
- Mostowy S, Tsolaki AG, Small PM, Behr MA. The in vitro evolution of BCG vaccines. Vaccine. 2003;21(30):4270-4274.
- Fleischmann RD, Alland D, Eisen JA, Carpenter L, White O, Peterson J, et al. Whole-genome comparison of Mycobacterium tuberculosis clinical and laboratory strains. J Bacterol. 2002;184(19):5479-5490.
- Pym AS, Brodin P, Brosch R, Huerre M, Cole ST. Loss of RD1 contributed to the attenuation of the live tuberculosis vaccines Mycobacterium bovis BCG and Mycobacterium microti. Mol Microbiol. 2002;46(3):709-717.
- Pizza M, Scarlato V, Masignani V, Giuliani MM, Arico B, Comanducci M, et al. Identification of vaccine candidates against serogroup B meningococcus by whole-genome sequencing. Science. 2000;287(5459):1816-1820.
- Giuliani MM, Adu-Bobie J, Comanducci M, Aricò B, Savino S, Santini L, et al. A universal vaccine for serogroup B meningococcus. Proc Natl Acad Sci. 2006;103(29):10834-10839
- Capecchi B, Adu‐Bobie J, Di Marcello F, Ciucchi L, Masignani V, Taddei A, et al. Neisseria meningitidis NadA is a new invasin which promotes bacterial adhesion to and penetration into human epithelial cells. Mol Microbiol. 2005;55(3):687-698.
- Madico G, Welsch JA, Lewis LA, McNaughton A, Perlman DH, Costello CE, et al. The meningococcal vaccine candidate GNA1870 binds the complement regulatory protein factor H and enhances serum resistance. J Immunol. 2006;177(1):501-510.
- Saha S, Raghava GP. Prediction of continuous B-cell epitopes in an antigen using recurrent neural network. Proteins: Structure, Function, and Bioinformatics. 2006 Oct 1;65(1):40-48.
- Serruto D, Rappuoli R. Post-genomic vaccine development. FEBS letters. 2006 ;580(12):2985-2992.
- Reche PA, Reinherz EL. PEPVAC: A web server for multi-epitope vaccine development based on the prediction of supertypic MHC ligands. Nucleic Acids Research. 2005 Jul 1;33(2):138-142.
- Mahan MJ, Tobias JW, Slauch JM, Hanna PC, Collier RJ, Mekalanos JJ. Antibiotic-based selection for bacterial genes that are specifically induced during infection of a host. Proc Natl Acad Sci U S A. 1995;92(3):669-673.
- Rappuoli R. Conjugates and reverse vaccinology to eliminate bacterial meningitis. Vaccine. 2001;19(19):2319-2322.
- Montigiani S, Falugi F, Scarselli M, Finco O, Petracca R, Galli G, et al. Genomic approach for analysis of surface proteins in Chlamydia pneumoniae. Infect Immun. 2002;70(1):368-379.
- Adu-Bobie J, Capecchi B, Serruto D, Rappuoli R, Pizza M. Two years into reverse vaccinology. Vaccine. 2003;21(7-8):605-610.
- Morgan AJ, Parker S. Translational mini review series on vaccines: the Edward Jenner Museum and the history of vaccination. Clin Exp Immunol. 2007;147(3):389-394.
- Dharmadi Y, Gonzalez R. DNA microarrays: Experimental issues, data analysis, and application to bacterial systems. Biotechnol Prog. 2004;20(5):1309-1324.
- He Y, Rappuoli R, De Groot, Chen RT. Emerging vaccine informatics. J Biomed Biotechnol. 2010;2010:218590 .
- Chiang SL, Mekalanos JJ, Holden DW. In vivo genetic analysis of bacterial virulence. Annu Rev Microbiol. 1999;53(1):129-154.
- Tang C, Holden D. Pathogen virulence genes-implications for vaccines and drug therapy. Br Med Bull. 1999;55(2):387-400.
- Lehoux DE, Levesque RC. Detection of genes essential in specific niches by signature-tagged mutagenesis. Curr Opin Biotechnol. 2000;11(5):434-439.
- Sun YH, Bakshi S, Chalmers R, Tang CM. Functional genomics of Neisseria meningitidis pathogenesis. Nat Med. 2000;6(11):1269-1273.
- Jones AL, Knoll KM, Rubens CE. Identification of Streptococcus agalactiae virulence genes in the neonatal rat sepsis model using signature‐tagged mutagenesis. Mol Microbiol. 2000;37(6):1444-1455
- Mahan MJ, Tobias JW, Slauch JM, Hanna PC, Collier RJ, Mekalanos JJ. Antibiotic-based selection for bacterial genes that are specifically induced during infection of a host. Proc Natl Acad Sci U S A. 1995;92(3):669-673.
- Valdivia RH, Hromockyj AE, Monack D, Ramakrishnan L, Falkow S. Applications for green fluorescent protein (GFP) in the study of hostpathogen interactions. Gene. 1996;173(1):47-52.
- Valdivia RH, Falkow S. Fluorescence-based isolation of bacterial genes expressed within host cells. Science. 1997;277(5334):2007-2011.
- Chiang SL, Mekalanos JJ, Holden DW. In vivo genetic analysis of bacterial virulence. Annu Rev Microbiol. 1999;53(1):129-154.
- Tang C, Holden D. Pathogen virulence genes-implications for vaccines and drug therapy. Br Med Bull. 1999;55(2):387-400.
- Coleman ML, Sullivan MB, Martiny AC, Steglich C, Barry K, DeLong EF, et al. Genomic islands and the ecology and evolution of Prochlorococcus. Science. 2006;311(5768):1768-1770.
- Rodriguez F., Martin-Cuadrado A, Rodriguez-Brito B, Pasic L, Thingstad TF. Explaining microbial population genomics through phage predation. Nat Rev Microbiol. 2009;7: 828-836.
- Pasic L, Rodriguez-Mueller B, Martin-Cuadrado A, Mira A, Rohwer F, Rodriguez Valera. Metagenomic islands of hyperhalophiles: the case of Salinibacter ruber. BMC Genomics.2009; 10(1): 570.
- Parker CE, Pearson TW, Anderson NL, Borchers CH. Mass-spectrometry-based clinical proteomics: A review and prospective. Analyst. 2010;135(8):1830-1838.
- Chakravarti DN, Fiske MJ, Fletcher LD, Zagursky RJ. Application of genomics and proteomics for identification of bacterial gene products as potential vaccine candidates. Vaccine. 2000;19(6):601-612.
- Wilkowsky SE, Farber M, Echaide I, De Echaide ST, Zamorano PI, Dominguez M, et al. Babesia bovis merozoite surface protein-2c (MSA-2c) contains highly immunogenic, conserved B-cell epitopes that elicit neutralization-sensitive antibodies in cattle. Mol Biochem Parasitol. 2000;127(2):133-141.
- Righetti PG, Hamdan M, Reymond F, Rossier JL. The proteome, anno domini two zero zero three. Genomics, Proteomics and Vaccines. 2004;103-134.
- Vytvytska O, Nagy E, Blüggel M, Meyer HE, Kurzbauer R, Huber LA. Identification of vaccine candidate antigens of Staphylococcus aureus by serological proteome analysis. Proteomics. 2002;2(5):580-590.
- Grandi G. Antibacterial vaccine design using genomics and proteomics. Trends Biotechnol. 2001;19(5):181-188.
- Hughes MJ, Moore JC, Lane JD, Wilson R, Pribul PK, Younes ZN, et al. Identification of major outer surface proteins of Streptococcus agalactiae. Infect. Immun. 2002.70: 1254-1259.
- Montigiani S, Falugi F, Scarselli M, Finco O, Petracca R, Galli G, et al. Genomic approach for analysis of surface proteins in Chlamydia pneumoniae. Infect Immun. 2002;70(1):368-379.
- Paustian ML, Kapur V, Bannantine JP. Comparative genomic hybridizations reveal genetic regions within the Mycobacterium avium complex that are divergent from Mycobacterium avium subsp. paratuberculosis isolates. J Bacteriol. 2005;187(7):2406-2415.
- Bloom BR, Murray CJ. Tuberculosis: Commentary on a reemergent killer. Science. 1992;257(5073):1055-1064.
- Lo M, Bulach DM, Powell DR, Haake DA, Matsunaga J, Paustian ML, et al. Effects of temperature on gene expression patterns in Leptospira interrogans serovar Lai as assessed by whole-genome microarrays. Infect Immun. 2006;74(10):5848-5859.
- Conway T, Schoolnik. Microarray expression profiling: Capturing a genome‐wide portrait of the transcriptome. Mol Microbiol. 2003;47(4):879-889.
- Bryant PA, Venter D, Robins-Browne R, Curtis N. Chips with everything: DNA microarrays in infectious diseases. Lancet Infect Dis. 2004;4(2):100-111.
- Rappuoli R. Pushing the limits of cellular microbiology: Microarrays to study bacteria-host cell intimate contacts. Proc Natl Acad. 2000;97(25):13467-13469
- Rappuoli R. Reverse vaccinology. Curr Opin Microbiol. 2000;3(5):445-450.
- Rappuoli R. Pushing the limits of cellular microbiology: microarrays to study bacteria–host cell intimate contacts. Proc Natl Acad. 2000 Dec 5;97(25):13467-13469.
- Grifantini R, Bartolini E, Muzzi A, Draghi M, Frigimelica E, Berger J, et al. Previously unrecognized vaccine candidates against group B meningococcus identified by DNA microarrays. Nat Biotechnol. 2002;20(9):914-921.
- Cummings CA, Relman DA. Using DNA microarrays to study host-microbe interactions. Emerg Infect Dis. 2000 Sep;6(5):513002E
- Kato‐Maeda M, Gao Q, Small PM. Microarray analysis of pathogens and their interaction with hosts: Technoreview. Cell Microbiol. 2001;3(11):713-719.
- Reche PA, Reinherz EL. PEPVAC: A web server for multi-epitope vaccine development based on the prediction of supertypic MHC ligands. Nucleic Acids Research. 2005 Jul 1;33(2):138-142.
- Office of International des Epizooties (OIE), No more deaths from rinderpest. 2011.
- Kurz S, Hübner C, Aepinus C, Theiss S, Guckenberger M, Panzner U, et al. Transcriptome-based antigen identification for Neisseria meningitidis. Vaccine. 2003;21(7-8):768-775.
- Gygi SP, Rochon Y, Franza BR, Aebersold R. Correlation between protein and mRNA abundance in yeast. Mol Cell Biol. 1999;19(3):1720-1730
- Tettelin H, Masignani V, Cieslewicz MJ, Eisen JA, Peterson S, Wessels MR, et al. Complete genome sequence and comparative genomic analysis of an emerging human pathogen, serotype V Streptococcus agalactiae. Proc. Natl. Acad. 2002;99(19):12391-12396.
- He Y, Rappuoli R, De Groot, Chen RT. Emerging vaccine informatics. J Biomed Biotechnol. 2010;2010:218590.
- Korber B, LaBute M, Yusim K. Immunoinformatics comes of age. PLoS Comput Biol. 2006;2(6):e71
- Zhang GL, Petrovsky N, Kwoh CK, August JT, Brusic V. PRED TAP: A system for prediction of peptide binding to the human transporter associated with antigen processing. Immunome Res. 2006;2(1):1-2.
- Nielsen M, Lundegaard C, Worning P, Hvid CS, Lamberth K, Buus S, et al. Improved prediction of MHC class I and class II epitopes using a novel Gibbs sampling approach. Bioinformatics. 2004;20(9):1388-1397.
- Brusic V, Bajic VB, Petrovsky N. Computational methods for prediction of T-cell epitopes-a framework for modelling, testing, and applications. Methods. 2004;34(4):436-443.
- Blythe MJ, Flower DR. Benchmarking B cell epitope prediction: underperformance of existing methods. Protein Sci. 2005;14(1):246-248.
- Saha S, Raghava GP. Prediction of continuous B-cell epitopes in an antigen using recurrent neural network. Proteins: Structure, Function, and Bioinformatics. 2006 Oct 1;65(1):40-48.
- Söllner J. Selection and combination of machine learning classifiers for prediction of linear B‐cell epitopes on proteins. J Mol Recognit. 2006;19(3):209-214.
- Partidos CD, Steward MW. Mimotopes of viral antigens and biologically important molecules as candidate vaccines and potential immunotherapeutics. Combinatorial chemistry & high throughput screening. 2002; 5(1):15-27.
- Caoili SE. A structural-energetic basis for B-cell epitope prediction. Protein Pept Lett. 2006;13(7):743-751.
- Haste Andersen P, Nielsen M, Lund OL. Prediction of residues in discontinuous B‐cell epitopes using protein 3D structures. Protein Sci. 2006;15(11):2558-2567
- Reche PA, Reinherz EL. Sequence variability analysis of human class I and class II MHC molecules: functional and structural correlates of amino acid polymorphisms. J Mol Biol. 2003.
- Reche PA, Reinherz EL. PEPVAC: A web server for multi-epitope vaccine development based on the prediction of supertypic MHC ligands. Nucleic Acids Research. 2005 Jul 1;33(2):138-142.
- Doytchinova IA, Guan P, Flower DR. Identifiying human MHC supertypes using bioinformatic methods. J Immunol. 2004;172(7):4314-4323.
- Gill SR, Pop M, DeBoy RT, Eckburg PB, Turnbaugh PJ, Samuel BS et al. Metagenomic analysis of the human distal gut microbiome. Science. 2006;312(5778):1355-1359.
- Kumánovics A, Takada T, Lindahl KF. Genomic organization of the mammalian MHC. Annu Rev Immunol. 2003;21(1):629-657.
- Kurokawa K, Itoh T, Kuwahara T, Oshima K, Toh H, Toyoda A, et al. Comparative metagenomics revealed commonly enriched gene sets in human gut microbiomes. DNA Res. 2007;14(4):169-181
Citation: Yohans H (2021) Review on Novel Bacterial Antigen Identification Methods for the Development of Candidate Vaccine. J Vaccines Vaccin. 12:465.
Copyright: © 2021 Yohans H. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.