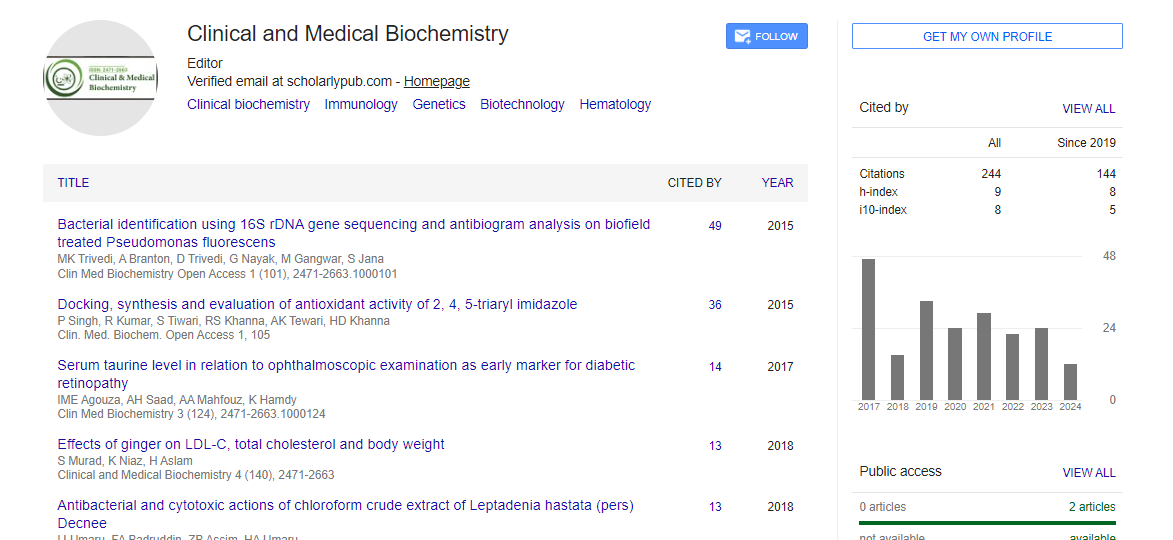

Indexed In
- RefSeek
- Directory of Research Journal Indexing (DRJI)
- Hamdard University
- EBSCO A-Z
- OCLC- WorldCat
- Scholarsteer
- Publons
- Euro Pub
- Google Scholar
Useful Links
Share This Page
Journal Flyer

Open Access Journals
- Agri and Aquaculture
- Biochemistry
- Bioinformatics & Systems Biology
- Business & Management
- Chemistry
- Clinical Sciences
- Engineering
- Food & Nutrition
- General Science
- Genetics & Molecular Biology
- Immunology & Microbiology
- Medical Sciences
- Neuroscience & Psychology
- Nursing & Health Care
- Pharmaceutical Sciences
Research Article - (2023) Volume 9, Issue 1
Quantitative Structure Activity Relationship and Molecular Docking Studies on a Series of Thiourea and Thiazolidine-4-Carboxylic Acid Analogues as Potent Neuraminidase Inhibitors
Abha Shrivastava1, Basheerulla Shaik2, Manish Rao Ambedkar3* and Vijay K Agrawal12Department of Applied Sciences, National Institute of Technical Teachers Training and Research Institute, Bhopal, India
3Department of Chemistry, MMH (PG) College, Ghaziabad, India
Received: 06-Sep-2022, Manuscript No. CMBO-22-17945; Editor assigned: 08-Sep-2022, Pre QC No. CMBO-22-17945 (PQ); Reviewed: 20-Sep-2022, QC No. CMBO-22-17945; Revised: 27-Jan-2023, Manuscript No. CMBO-22-17945 (R); Published: 03-Feb-2023, DOI: 10.35841/2471-2663.23.9.148
Abstract
Neuraminidase inhibitors are a type of medication that inhibits the neuraminidase enzyme. They are type of antiviral medication that is often used to treat influenza. In this paper we have taken 53 compounds which are derivatives of thiourea and thiazolidine-4-carboxylic acid derivatives. We modelled the pIC50 activity using RDF115p, E2s, R1i parameters. The excellent value of r2=0.725 shows that following model is best suitable. We also performed docking study and the best docking score is -28.1891, and the best predicted activity is 8.36.
Keywords
QSAR studies; Docking; Neuraminidase inhibitors; Thiourea; pIC50 activity
Introduction
Neuraminidase (NA) is certainly considered one among glycoproteins on the floor of the influenza virus [1]. NA is accountable for viral launch from inflamed cells and viral shipping through the mucus with inside the breathing tract. Na has been identified as an ability goal for the manage influenza virus [2].
Neuraminidase Inhibitors (NIs) from key additives of pandemic preparedness plans as remedy and prophylaxis may want to lessen virus transmission [3]. The sialic acid analogs were the first NIs reported. Based on the structure of sialic acid, different NI series have been prepared, such as cyclohexenes, benzoic acids, pyrolidine derivatives etc. [4-7].
In the past decade, thiourea derivatives have been reported to be effective against HIV and have bactericidal effects [8]. Neuraminidase stays an appealing anti-influenza drug target, at the same time as the emergence of viruses proof against the presently to be had drug has offered a brand new challenge. Noticeably, the crystal shape of the organization-1 neuraminidase (H1,N4 and N8) found out a unique hollow space adjoining to the lively web website online in organization 1 however now no longer in organization 2 proteins crystallography, suggesting new possibilities for drug layout that concentrate on this hollow space similarly to acknowledged lively web website online [9].
Earlier crystallographic and making sure SAR research have found out that the lively web page of NA can be divided into 4 important binding sites. Steindl and Lange defined the improvement of surprisingly selective pharmacophore fashions for inhibitors of viral NA inside the catalyst section possess sturdy structural resemblance in the ones parts, which correspond to the truth that the 4 wallet are important for interplay with the lively web page of NA [10].
However, few studies have evaluated substituted acyl (thio) urea and 2H-1,2,4-thiadiazolo (2,3-α) pyrimidines for their antiviral activities. In 2006, a brand new elegance of substituted acyl (thio) urea and 2H-1,2,4-thiadiazolo (2,3-α) pyrimidine derivatives with notably particular anti-influenza virus class have been organized with the aid of using Sun, et al. Their in vitro inhibitory class of opposition to influenza neuraminidase (H1N1) has been additionally investigated and located to correlate nicely with their antiviral efficacy in class of subculture.
Studied QSAR of forty thiourea analogs the use of spatial, topological, electronic, thermodynamic and E-state indices. Yu Liu, et al. have derived that thiazolidine-4-carboxylic acid could show potent NA inhibitory activity and his finding can be used to design novel influenza NA inhibitors that exhibit increased activity based on Thiazolidine ring [11]. Also, Thiazolidine-4- carboxylic acid was synthesized in good yields starting from commercially available L-cysteine hydrochloride using a suitable synthetic strategy.
Materials and Methods
All the compounds for the study have been taken from Jiaying sun, et al. and Yu liu, et al. All the compounds are listed in Table 1 along with their inhibitory activity. For QSAR studies, out of 53 compounds, 75% of them (40 compounds) were selected for the training set by random selection, and the remaining 25% (13 compounds) were used for the test set for evaluating the predictability of the developed models.
| Compound no. | Molecular structure | RDF115p | E2s | R1i | pIC50 Obsd. |
pIC50 Cald. From eq. 1 |
ΔpIC50 |
|---|---|---|---|---|---|---|---|
| 1* |  |
0.073 | 0.418 | 2.568 | 5.78 | 6.110 | 0.327 |
| 2 |  |
3.305 | 0.550 | 2.553 | 7.1 | 6.915 | -0.182 |
| 3 |  |
0.000 | 0.482 | 2.575 | 6.49 | 6.205 | -0.290 |
| 4 |  |
0.000 | 0.415 | 2.605 | 5.75 | 6.219 | 0.467 |
| 5 |  |
0.000 | 0.538 | 2.330 | 4.84 | 5.434 | 0.595 |
| 6 |  |
0.000 | 0.344 | 2.406 | 5.78 | 5.436 | -0.344 |
| 7 |  |
0.187 | 0.324 | 2.640 | 5.64 | 6.257 | 0.619 |
| 8 |  |
0.000 | 0.388 | 2.653 | 6.44 | 6.348 | -0.096 |
| 9 |  |
0.068 | 0.456 | 2.399 | 5.85 | 5.577 | -0.271 |
| 10 |  |
0.000 | 0.374 | 2.537 | 5.89 | 5.929 | 0.043 |
| 11 |  |
0.000 | 0.584 | 2.384 | 5.75 | 5.683 | -0.064 |
| 12* |  |
0.528 | 0.552 | 2.171 | 5.74 | 5.015 | -0.723 |
| 13 |  |
2.112 | 0.677 | 2.242 | 5.78 | 5.761 | -0.016 |
| 14* |  |
0.085 | 0.301 | 2.320 | 5.84 | 5.100 | -0.745 |
| 15* |  |
0.000 | 0.238 | 2.647 | 5.91 | 6.126 | 0.212 |
| 16 |  |
0.000 | 0.502 | 2.517 | 5.89 | 6.032 | 0.143 |
| 17 |  |
0.000 | 0.403 | 2.426 | 5.07 | 5.585 | 0.518 |
| 18 |  |
0.205 | 0.210 | 2.388 | 5.14 | 5.238 | 0.095 |
| 19 |  |
0.336 | 0.450 | 2.283 | 5.59 | 5.225 | -0.362 |
| 20 |  |
0.668 | 0.428 | 2.362 | 4.73 | 5.538 | 0.805 |
| 21 |  |
0.223 | 0.477 | 2.403 | 5.68 | 5.651 | -0.027 |
| 22* |  |
0.472 | 0.323 | 2.491 | 6.51 | 5.801 | -0.708 |
| 23 |  |
0.101 | 0.620 | 2.313 | 5.87 | 5.507 | -0.359 |
| 24 |  |
0.000 | 0.425 | 2.251 | 5.29 | 5.010 | -0.282 |
| 25 |  |
0.723 | 0.663 | 2.379 | 5.72 | 5.923 | 0.199 |
| 26 |  |
0.000 | 0.263 | 2.168 | 4.67 | 4.506 | -0.166 |
| 27 |  |
0.000 | 0.295 | 2.233 | 4.69 | 4.774 | 0.079 |
| 28 |  |
0.000 | 0.352 | 2.286 | 4.74 | 5.033 | 0.291 |
| 29* |  |
0.000 | 0.226 | 2.140 | 4.63 | 4.360 | -0.271 |
| 30 |  |
0.000 | 0.386 | 2.216 | 4.65 | 4.837 | 0.189 |
| 31* |  |
0.000 | 0.210 | 2.297 | 4.91 | 4.880 | -0.030 |
| 32 |  |
0.000 | 0.300 | 2.179 | 4.37 | 4.594 | 0.228 |
| 33 |  |
0.000 | 0.708 | 2.160 | 5.12 | 5.076 | -0.047 |
| 34 |  |
0.000 | 0.633 | 2.224 | 5.23 | 5.196 | -0.038 |
| 35 |  |
0.000 | 0.805 | 2.282 | 4.97 | 5.627 | 0.656 |
| 36 |  |
0.000 | 0.575 | 2.133 | 5.06 | 4.804 | -0.259 |
| 37 |  |
0.000 | 0.401 | 2.222 | 5.12 | 4.878 | -0.238 |
| 38 |  |
0.000 | 0.635 | 2.298 | 5.10 | 5.454 | 0.353 |
| 39* |  |
0.000 | 0.715 | 2.155 | 4.89 | 5.068 | 0.179 |
| 40* |  |
0.000 | 0.414 | 2.407 | 5.92 | 5.534 | -0.383 |
| 41* |  |
0.000 | 0.301 | 2.451 | 6.19 | 5.534 | -0.653 |
| 42 |  |
0.000 | 0.252 | 2.457 | 5.72 | 5.489 | -0.228 |
| 43 |  |
0.000 | 0.292 | 2.391 | 5.61 | 5.315 | -0.292 |
| 44* |  |
0.000 | 0.243 | 2.448 | 5.73 | 5.446 | -0.282 |
| 45 |  |
0.000 | 0.285 | 2.509 | 5.79 | 5.713 | -0.077 |
| 46 |  |
0.000 | 0.304 | 2.401 | 5.54 | 5.366 | -0.174 |
| 47 |  |
0.000 | 0.706 | 2.438 | 6.28 | 6.033 | -0.243 |
| 48 |  |
0.000 | 0.614 | 2.507 | 6.68 | 6.147 | -0.531 |
| 49 |  |
0.000 | 0.551 | 2.559 | 6.55 | 6.242 | -0.311 |
| 50 |  |
0.000 | 0.542 | 2.414 | 6.09 | 5.730 | -0.362 |
| 51* |  |
0.000 | 0.478 | 2.484 | 5.99 | 5.886 | -0.106 |
| 52* |  |
0.000 | 0.558 | 2.580 | 6.85 | 6.324 | -0.530 |
| 53 |  |
0.000 | 0.674 | 2.438 | 6.01 | 5.990 | -0.019 |
Table 1: A series thiourea analogs and thiazolidine-4-carboxylic acid derivatives as potent influenza neuraminidase inhibitors.
The chemical structure was drawn using ACD/Chemskecth software and the physiochemical/topological descriptors were calculated using Alva software [12]. Among all the calculated descriptors, descriptors listed in Table 2 are found to be correlated with the activity.
| Compoundno. | Structure | RDF115p | E2s | R1i | pIC50 |
|---|---|---|---|---|---|
| 1 |  |
7.376 | 0.667 | 2.678 | 8.36 |
| 2 |  |
8.126 | 0.467 | 2.619 | 8.04 |
| 3 |  |
4.979 | 0.547 | 2.701 | 7.77 |
| 4 |  |
2.883 | 0.6 | 2.63 | 7.16 |
| 5 |  |
4.856 | 0.514 | 2.629 | 7.45 |
| 6 |  |
5.964 | 0.487 | 2.627 | 7.64 |
| 7 |  |
3.262 | 0.536 | 2.674 | 7.3 |
| 8 |  |
3.531 | 0.554 | 2.706 | 7.5 |
| 9 |  |
6.25 | 0.469 | 2.636 | 7.71 |
| 10 |  |
3.957 | 0.606 | 2.761 | 7.85 |
Table 2: Predicted compounds with proposed activity values.
In the Table 1, the test set compounds are marked with superscript (*). The most significant structural descriptors that were found to govern the activity of the compounds were as follows.
• RDF115p
• E2s
• R1i
Results and Discussion
An Multiple Linear Regression (MLR) analysis was performed using statistical data miner on the training set compounds to establish a correlation between activity and various descriptors of the compounds. The most significant correlation obtained is shown by equation 1.
pIC50=-3.3304+0.2102 (± 0.1842) RDF115p+1.3421 (± 0.7593) E2s+3.4518 (± 0.8113) R1i …….………………………… (1)
n=40, r2=0.7249, r2cv=0.655, r2 pred=0.652, s=0.343, F=31.617
In equation 1, n refers to the number of data points used in the correlation, r2 is the square of the correlation coefficient, r2 cv is the square of cross validated correlation coefficient obtained by Leave One Out (LOO) jackknife procedure, and r2 pred is the square of correlation coefficient obtained for test set compounds to judge the external validity of the correlation.
Values of r2cv and r2 pred are calculated according to equation 2 and 3, respectively, where obsd in equation 2 refers to the observed activity of compound in the training set and that in equation 3 to the compound obsd in test set. Similarly, pred in equation 2 refers to the predicted activity of compound pred in the training set obtained in LOO jackknife procedure and that in equation 3 to that predicted for the test test compounds by model obtained for the training set. However, av, obsd in both the equation refers to the average activity of the training set compound.
r2cv=1−(Σ (obsd−pred)2/Σ (obsd–av, obsd)2) …………………… (2)
The correlation is supposed to be valid and has the good internal predictive ability if r2cv>0.60. Similarly, the external predictive ability of the model is supposed to be good if its r2pred>0.5. From both the parameters, the correlation expressed by equation 1 is found to be quite valid. Among the remaining two statistical parameters, s and F, s is the standard deviation and F is the Fischer ratio between the variances of calculated and observed activities. The figure within the parentheses with (±) sign refers to the 95% confidence intervals.
The F-value given in parenthesis refers to the standard F value at the 99% level. A higher value of F indicates a good correlation. Also, all the descriptors used in this correlation are found to be quite significant if we remove them one by one, the significance of the correlation is appreciably dropped (equation 4 and 5).
PIC50=-3.628+1.525( ± 0.783) E2s+3.558 (± 0.851) R1i …… (4)
n=40, r2=0.684, r2cv=0.575, r2 pred=0.639, s=0.363, F=40.034
PIC50=-2.406+3.347(± 0.992) R1i …….………………………… (5)
n=40, r2=0.551, r2cv=0.497, r2pred=0.543, s=0.426, F=46.663
Thus, from the above results, it is clear that equation 1 has a noteworthy correlation between the inhibitory activity and the structural descriptors of the compounds. Although the correlation does not have any mechanistic aspects, but it has good predictive ability. A graph drawn between the predicated and observed activities for both the training and test sets further shows that the model has good predictive ability. Figure 1 shows that except 1 or 2 points, all other points lie near the straight line. Using this MLR model (equation 1), we have predicted some new compounds, as shown in Table 2, where each compound has a higher activity value than any compound in the existing series.

Figure 1:A plot between predicted and observed activities of compounds of Table 1.
Docking analysis
Molecular docking was performed on the predicted compounds in Table 2 using lead IT Flexx software to get the binding mode of these compounds. The potency of a molecule is determined by its ability to interact with an enzyme. For studying molecular docking, the crystal structure of the related enzyme is very important, which can now be retrieved from the RCSB protein data bank. We selected the enzyme with PDB entry code 1A4G.
The compounds listed in Table 2 were docked into this enzyme and their docking results are shown in Table 3. The molecular docking analysis was carried out on all of the compounds predicted to be present in the enzyme.
| Compoundno. | No. of H-bonds | H-bonds | H-bonds length | Score |
| 1 | 5 | O(11)-LYS44 O(27)-ASN150 H(38)-MET96 H(49)-GLY27 H(51)-THR23 |
4.70 4.61 3.35 4.47 4.38 |
-27.3170 |
| 2 | 5 | O(11)-LYS44 O(27)-ASN150 H(39)-MET96 H(50)-GLY27 H(52)-THR23 |
4.70 4.60 3.35 4.34 4.36 |
-27.9540 |
| 3 | 4 | O(25)-ASP103 H(52)-ASP103 H(52)-ASP103 H(53)-LEU21 |
3.51 8.16 3.36 4.43 |
-27.1239 |
| 4 | 10 | N(9)-ARG118 O(11)-TYR406 O(14)-ARG292 O(14)-ARG292 O(14)-ARG371 O(25)-ASN294 O(25)-ARG292 H(35)-GLU119 H(46)-GLU276 H(57)-GLU276 |
0.51 2.51 2.85 3.96 4.70 2.61 0.32 4.70 0.21 0.43 |
-20.5896 |
| 5 | 5 | O(11)-ARG371 O(11)-ARG118 O(14)-GLY348 O(25)-ARG152 H(48)-GLU152 |
4.19 4.7 4.28 4.38 8.3 |
-19.5077 |
| 6 | 5 | O(11)-ARG118 O(11)-ARG371 O(14)-GLY348 O(25)-ARG152 H(49)-GLU119 |
4.7 4.21 4.28 4.37 8.3 |
-18.6483 |
| 7 | 9 | O(14)-ARG292 O(14)-ARG292 O(14)-ARG371 O(25)-ARG152 O(27)-ARG156 H(39)-ASP151 H(51)-GLU119 H(53)-ASP178 H(72)-GLU119 |
2.36 3.17 4.7 4.7 3.03 2.73 8.3 1.25 0.13 |
-23.0806 |
| 8 | 7 | O(14)-ARG371 O(14)-ARG118 O(25)-ARG152 O(27)-ARG156 H(39)-ASP151 H(51)-GLU119 H(53)-TRP178 |
2.34 4.7 4.7 3.21 1.28 8.3 1.88 |
-24.5201 |
| 9 | 5 | O(11)-ARG118 O(11)-ARG371 O(14)-GLY348 O(25)-ARG152 H(50)-GLU119 |
4.7 2.97 3.41 0.26 8.3 |
-21.3755 |
| 10 | 6 | O(14)-ASP103 N(29)-THR23 H(39)-ASP103 H(39)-ASP103 H(51)-GLU149 H(57)-THR23 |
4.54 3.74 2.27 0.59 4.7 4.24 |
-28.1891 |
Table 3: Molecular docking results of the predicted molecules.
Here we cited only compound 1, this compound having the highest predicted activity and the compound 10 having the highest docking score just to illustrate the best possible interactions between the inhibitors and the enzyme (Figure 1). From Figures 2 and 3 it is clear that the predicted compounds have good interactions with the enzyme. They all undergo hydrogen bondings as well as steric interactions, in which several moieties of compounds are surrounded by the different active clefts of the enzyme (Table 4). The penetration of any moiety of any inhibitor in any cavity of the enzyme will depend on its flexibility. All these steric interactions might involve dispersion interactions, which is a set of electronic interactions.
| Total Mol weight | cLogP | cLogS | H-Acceptors | H-Donors |
|---|---|---|---|---|
| 490.971 | 1.5781 | -4.484 | 10 | 4 |
| 513.061 | 2.7389 | -4.805 | 10 | 4 |
| 527.088 | 3.1933 | -5.075 | 10 | 4 |
| 527.088 | 3.1933 | -5.075 | 10 | 4 |
| 561.105 | 3.1769 | -5.508 | 10 | 4 |
| 567.153 | 3.5335 | -5.969 | 10 | 4 |
| 533.051 | 2.3632 | -4.86 | 10 | 4 |
| 540.954 | -0.4975 | -4.065 | 12 | 5 |
| 521.001 | -0.325 | -4.084 | 12 | 5 |
| 529.064 | 0.3814 | -4.135 | 12 | 5 |
| 591.135 | 1.2738 | -5.108 | 12 | 5 |
Table 4: Pharmacokinetic properties of the predicted compounds of Table 2.

Figure 2:A representation of the binding of predicted compound 1 (Table 2) in 1A4G (PDBID: 1A4G).

Figure 3:A representation of the binding of predicted compound 10 (Table 2) in 1A4G (PDBID: 1A4G).
Pharmacokinetic studies
The pharmacokinetic properties of the predicted compounds were obtained using data warriors software, and the results are shown in Table 3. These pharmacokinetic properties include Molecular Weight (MW), ClogP, number of H-bond Acceptors (HA), and number of H-bond Donors (HD). According to Lipinski's rule of five, compounds with MW<500 and ClogP<5 should have good absorption and penetration capacity.
Conclusion
The inhibition activity of a series of thiourea analogs compounds was found to be well correlated with various physicochemical properties. The correlation between activity and structure provided new information about which thiourea based compounds are more active. Through docking studies of the predicted compounds, it was found that all of them interact with the enzyme 1A4G in a number of ways, involving their bulky groups in significant steric interactions with some of the site's residues. Studies on the pharmacokinetic characteristics of the predicted compounds show that they have good pharmacokinetic properties.
References
- McKimm-Breschkin JL. Resistance of influenza viruses to neuraminidase inhibitors-A review. Antiviral Res. 2000;47(1):1-7.
- Verma RP, Hansch C. A QSAR study on influenza neuraminidase inhibitors. Bioorg Med Chem. 2006;14(4):982-996.
- Stephenson I, Clark TW, Pareek M. Antiviral treatment and prevention of seasonal influenza: A comparative review of recommendations in the European Union. J Clin Virol. 2008;42(3):244-248.
- Kim CU, Lew W, Williams MA, Wu H, Zhang L, Chen X, et al. Structure Activity relationship studies of novel carbocyclic influenza neuraminidase inhibitors. J Med Chem. 1998;41(14):2451-2460.
- Brouillette WJ, Atigadda VR, Luo M, Air GM, Babu YS, Bantia S. Design of benzoic acid inhibitors of influenza neuraminidase containing a cyclic substitution for the N-acetyl grouping. Bioorg Med Chem Lett. 1999;9(14):1901-1906.
- Wang GT, Chen Y, Wang S, Gentles R, Sowin T, Kati W, et al. Design, synthesis, and structural analysis of influenza neuraminidase inhibitors containing pyrrolidine cores. J Med Chem. 2001;44(8):1192-1201.
- Krueger AC, Xu Y, Kati WM, Kempf DJ, Maring CJ, McDaniel KF, et al. Synthesis of potent pyrrolidine influenza neuraminidase inhibitors. Bioorg Med Chem Lett. 2008;18(5):1692-1695.
- Sun C, Zhang X, Huang H, Zhou P. Synthesis and evaluation of a new series of substituted acyl (thio) urea and thiadiazolo [2, 3-a] pyrimidine derivatives as potent inhibitors of influenza virus neuraminidase. Bioorg Med Chem. 2006;14(24):8574-8581.
- Russell RJ, Haire LF, Stevens DJ, Collins PJ, Lin YP, Blackburn GM, et al. The structure of H5N1 avian influenza neuraminidase suggests new opportunities for drug design. Nature. 2006;443(7107):45-49.
- Steindl T, Langer T. Influenza virus neuraminidase inhibitors: Generation and comparison of structure-based and common feature pharmacophore hypotheses and their application in virtual screening. J Chem Inf Comput Sci. 2004;44(5):1849-1856.
- Liu Y, Jing F, Xu Y, Xie Y, Shi F, Fang H, et al. Design, synthesis and biological activity of thiazolidine-4-carboxylic acid derivatives as novel influenza neuraminidase inhibitors. Bioorg Med Che. 2011;19(7):2342-2348.
- Sander T, Freyss J, von Korff M, Rufener C. DataWarrior: An open source program for chemistry aware data visualization and analysis. J Chem Inf Model. 2015;55(2):460-473.
Citation: Shrivastava A, Shaik B, Ambedkar MR, Agrawal VK (2023) Quantitative Structure Activity Relationship and Molecular Docking Studies on a Series of Thiourea and Thiazolidine-4-Carboxylic Acid Analogues as Potent Neuraminidase Inhibitors. Clin Med Bio Chem. 09:148.
Copyright: © 2023 Shrivastava A, et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.