


Indexed In
- Academic Journals Database
- Open J Gate
- Genamics JournalSeek
- JournalTOCs
- ResearchBible
- Ulrich's Periodicals Directory
- Electronic Journals Library
- RefSeek
- Hamdard University
- EBSCO A-Z
- OCLC- WorldCat
- Scholarsteer
- SWB online catalog
- Virtual Library of Biology (vifabio)
- Publons
- MIAR
- Geneva Foundation for Medical Education and Research
- Euro Pub
- Google Scholar
Useful Links
Share This Page
Journal Flyer

Open Access Journals
- Agri and Aquaculture
- Biochemistry
- Bioinformatics & Systems Biology
- Business & Management
- Chemistry
- Clinical Sciences
- Engineering
- Food & Nutrition
- General Science
- Genetics & Molecular Biology
- Immunology & Microbiology
- Medical Sciences
- Neuroscience & Psychology
- Nursing & Health Care
- Pharmaceutical Sciences
Research Article - (2022) Volume 13, Issue 1
Predictive Functionality of Bacteria in Naturally Fermented Milk Products of India Using PICRUSt2 and Piphillin Pipelines
H. Nakibapher Jones Shangpliang and Jyoti Prakash Tamang*Received: 03-Jan-2022, Manuscript No. JDMGP-22-15356; Editor assigned: 05-Jan-2022, Pre QC No. JDMGP-22-15356; Reviewed: 17-Jan-2022, QC No. JDMGP-22-15356; Revised: 20-Jan-2022, Manuscript No. JDMGP-22-15356; Published: 24-Jan-2022, DOI: 10.4172/2153-0602.22.13.244
Abstract
Naturally Fermented Milk (NFM) products are popular food delicacies in Indian states of Sikkim and Arunachal Pradesh. Bacterial communities in these NFM products of India were previously analysed by high-throughput sequence method. However, predictive gene functionality of NFM products of India has not been studied. In this study, raw sequences of NFM products of Sikkim and Arunachal Pradesh were accessed from MG-RAST/NCBI database server. PICRUSt2 and Piphillin tools were applied to study microbial functional gene prediction. MUSiCC- normalized KOs and mapped KEGG pathways from both PICRUSt2 and Piphillin resulted in higher percentage of the former in comparison to the latter. Though, functional features were compared from both the pipelines, however, there were significant differences between the predictions. Therefore, a consolidated presentation of both the algorithms presented an overall outlook into the predictive functional profiles associated with the microbiota of the NFM products of India.
Keywords
Metagenome gene prediction; PICRUSt2; Piphillin; Naturally fermented milk products; Lactic acid bacteria
Introduction
Naturally Fermented Milk (NFM) products are popular food items in daily diets of ethnic people of Arunachal Pradesh and Sikkim in India, which include dahi, mohi, gheu, soft-chhurpi, hard-chhurpi, dudh- chhurpi, chhu, somar, maa, philu, shyow, mar, chhurpi/churapi, churkam and churtang/chhurpupu [1,2]. Previously, taxonomic analysis using High-Throughput Sequencing (HTS) of NFM products of Arunachal Pradesh and Sikkim viz. chhurpi, churkam mar/gheu and dahi, have been studied [3]. We have recorded the abundance of phylum Firmicutes with predominated species of Lactic Acid Bacteria (LAB) viz. Lactococcus lactis (19.7%) and Lactobacillus helveticus (9.6%) and Leuconostoc mesenteroides (4.5%) and Acetic Acid Bacteria (AAB): Acetobacter lovaniensis (5.8%), Acetobacter pasteurianus (5.7%), Gluconobacter oxydans (5.3%), and Acetobacter syzygii (4.8%) [3]. Application of shotgun metagenomics is one of the commonly used methods for understanding the microbial- associated gene functional characteristics [4]. However, alternately functional profiles of a microbial community can also be inferred indirectly by marker-gene surveys such as 16S rRNA gene [5,6]. Bioinformatics pipelines such as Phylogenetic Investigation of Communities by Reconstruction of Unobserved States version2 (PICRUSt2) [7] and Piphillin [8] among others are some of the well-known tools for microbial predictive functionality studies from various NGS-related metagenomic data [5,6]. These pipelines have also been applied in fermented milk products to infer the functional gene predictions [9-11]. Microbiota present in NFM products harbour probiotic properties and impart several health-promoting benefits to consumers [12,13,14]. Predictive gene functionality in NFM products of India has not been analysed yet. Hence, the present study is aimed to predict the microbial functional contents of 16S rRNA gene sequencing data of NFM products of India, previously analysed by high-throughput sequencing method [3], using PICRUSt2 and Piphillin pipelines.
Material and Methods
Pre-analysis prior to predictive functionality analysis
Raw sequences of NFM products of Arunachal Pradesh and Sikkim in India analysed by HTS method [3] were accessed from MG-RAST/NCBI database server and were used in this study. Raw reads were processed using QIIME2-2020.6 (https://docs.qiime2.org/2020.6/) [15]. After importing into QIIME2 environment, Q-score based filtering and denoising was performed using Divisive Amplicon Denoising Algorithm (DADA2) [16] via qiime dada2 denoise-paired plugin. Quality-filtered sequences were then clustered against SILVA v132 [17] database and followed by taxonomic assignment using q2-vsearch-cluster-features-closed- reference [18]. Taxonomic assigned of the ASVs was acheived using SILVA v132 [17] database and relative abundance of the predominant genera was calculated (Table 1).
| Genus | Region | Relative percentage of abundance (%) | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Lactococcus | Lactobacillus | Acetobacter | Leuconostoc | Pseudomonas | Staphylococcus | Gluconobacter | Bacillus | Acinetobacter | Enterococcus | Others (<1%) | ||
| Chhurpi | Sikkim | 30.54 | 19.18 | 15.9 | 12.7 | 6.04 | 4.06 | 2.45 | 1.66 | 0.71 | 1.28 | 5.48 |
| Gheu | Sikkim | 28.92 | 4.43 | 4.51 | 4 | 49.81 | 0.45 | 1.13 | 0.24 | 2.15 | 0.21 | 4.15 |
| Dahi | Sikkim | 42.29 | 13.49 | 9.32 | 17.68 | 9.57 | 0.86 | 2.56 | 0.55 | 1.59 | 0.19 | 1.9 |
| Chhurpi | Arunachal Pradesh | 51.06 | 15.8 | 10.87 | 5.1 | 3.88 | 4.43 | 2.27 | 2.26 | 0.77 | 0.9 | 2.66 |
| Churkam | Arunachal Pradesh | 38.98 | 14.81 | 11.95 | 5.05 | 3.46 | 13.16 | 2.36 | 3.16 | 1.29 | 1.38 | 4.4 |
| Mar | Arunachal Pradesh | 35.22 | 14.12 | 19.22 | 8.65 | 4.56 | 4.65 | 7.11 | 2.56 | 0.82 | 0.98 | 2.11 |
| Average (%) | 37.84 | 13.64 | 11.96 | 8.86 | 12.89 | 4.6 | 2.98 | 1.74 | 1.22 | 0.82 | 3.45 | |
Table 1: Bacterial genera identified from different NFM products of Sikkim and Arunachal Pradesh.
Predictive functionality analysis
PICRUSt2 analysis (https://github.com/picrust/picrust2/wiki): PICRUSt2 is a pipeline for predicting functional abundances based only on marker gene sequences, 16S rRNA gene [7]. Here, the quality-filtered sequences were fed into PICRUSt2 algorithm with the default parameters. The representative sequences were first clustered in QIIME2 against SILVA v132 database [17]. Functional prediction in PICRUSt2 involves three main steps- phylogenetic placement of reads, hidden state prediction, pathway inference. Firstly, for phylogenetic placement of reads, multiple assignment of the Exact Sequence Variants (ESVs) was performed using HMMER (http://www.hmmer.org/); and placements of the ESVs in the reference tree was performed using Evolutionary Placement-ng (EPA-ng) [19] and Genesis Applications for Phylogenetic Placement Analyses (GAPPA) [20]. Secondly, hidden state prediction of the gene families was run by castor R package [21] with the default “maximum parsimony” algorithm. Lastly, for pathway inference, a modified version of MinPath packaged within PICRUSt2 is used and metagenome prediction was achieved using the default “metagenome_pipeline.py” script [22]. The output features were then mapped against KEGG (Kyoto Encyclopaedia of Genes and Genomes) database for systematic analysis of gene functions [23]. Furthermore, the KEGG pathway information was then collapsed into three different levels- Category (Level-1), Super Pathway (Level-2) and Pathways (Level-3).
Piphillin analysis (https://piphillin.secondgenome.com/): Alternatively, functional prediction was also performed using Piphillin software [8]. Piphillin is a straightforward independent algorithm which predicts gene functionality from the structural 16S rRNA gene without the use of any proposed phylogenetic tree unlike PICRUSt or PICRUSt2 [24]. Most importantly, Piphillin is a web-based analysis software which is simplified, user-friendly and has been shown to have better accuracy in predicting genome function from 16S rRNA gene content [8]. Piphillin uses KEGG and BioCyc database as reference databases. Here, gene copy numbers within each genome were retrieved and formatted by KO. Inference of gene function or metagenomic content was achieved by simply matching each representative of OTU/ASVs directly to the nearest sequenced genome without placing the sequence on the phylogenetic tree [24]. The representative OTU abundance table is then transformed into genome abundance table by using USEARCH with global alignment (Edgar 2010) and the resulting closest matched genome to the 16S rRNA gene copy of each representative OTUs/ASVs above identity threshold is considered as the inferred genome for each OTU/ASV (Iwai et al. 2016). In Piphillin analysis, DADA2-clustered representative sequences (.fasta) and ASV abundance frequency table (.csv) were required to upload to the Piphillin server (https://piphillin.secondgenome.com/) via a web-browser. Inferred metagenomic content output was then collapsed into three different levels- Category (Level-1), Super Pathway (Level-2) and Pathways (Level-3).
Statistical analysis and data visualization
Unnormalized Kyoto Encyclopaedia of Genes and Genomes (KEGG) ortholog (KO) profiles of PICRUSt2 and Piphillin predictive were normalized using Metagenomic Universal Single- Copy Correction (MUSiCC), a normalization paradigm which combines universal single-copy genes with machine learning tools to correct biases and to obtain accurate biological measure of gene abundance in metagenomic studies [25]. The output features were then mapped to KEGG database for systematic analysis of gene functions [23]. Error-corrected functional abundance table was then used for downstream analysis and rleative abundances (%) was plotted in MS-Excel v365 as stacked bar-plot for both PICRUSt2 and Piphillin predictive outputs. To check the significant differences between the functional content as predicted by both PICRSUt2 and Piphillin, White’s non-parametric with Benjamini- Hochberg FDR (false discovery rate) was applied in STAMP- statistical analysis of taxonomic and functional profiles [26] and visualized as extended error-bar chart with alpha significance of 0.05 (q-value) for all the functional levels – category (level-1), super pathway (level-2) and pathway (level-3). Furthermore, relationship between bacteria (lactic acid bacteria, LAB; acetic acid bacteria, AAB; and non-LAB/AAB) and functionalities were analyzed using non-parametric Spearman’s correlation in Statistical Package for the Social Sciences (SPSS) v20 and the correlation matrix was visualized as heatmap using ClustVis [27]. Significant interaction between bacteria and function are denoted with “*” <0.05 and “**” <0.01.
Results
Microbial predictive gene functionality
A total of 1109 error-corrected ESVs was obtained from DADA2 analysis and about 268 SILVA-clustered sequences were used for the downstream predictive analysis. A total of 5995 MUSiCC- normalized KOs and 181 mapped KEGG pathways was obtained from PICRUSt2 analysis. Similarly, a total of 5245 MUSiCC- normalized KOs and 157 mapped KEGG pathways was obtained from Piphillin analysis. Overall, both PICRUSt2 and Piphillin pipelines showed a similar pattern (Figure 1), except in the metabolism category where the PICRUSt2 was significantly higher in comparison to that predicted by Piphillin pipeline (Figure 2). Additionally, at the super pathway level, PICRUSt2 prediction showed significantly high in amino acid metabolism, metabolism of cofactors and vitamins, energy metabolism, and biosynthesis of other secondary metabolites (Figure 2). On the other hand, predictive super pathways which included carbohydrate metabolism, xenobiotics biodegradation and metabolism, metabolism of other amino acids, lipid metabolism, metabolism of terpenoids and polyketides, glycan biosynthesis and metabolism, and nucleotide metabolism were significantly higher through Piphillin prediction (Figure 2). Significant metabolic-related pathways inferred by both PICRUSt2 and Piphillin tools were compared showing several functional features predicted by these two pipelines (Figure 3).

Figure 1: An overall categorical representation of the MUSiCC-normalized predictive microbial functions as inferred by (a) PICRUSt2 and (b)
Piphillin. Note: (a) PICRUSt2; KEGG Pathways Relative abundance; 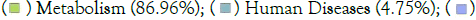 Environmental Information Processing (3.19%);
Environmental Information Processing (3.19%); 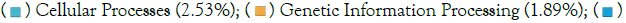 Organismal Systems (0.68%); (b) Piphillin; KEGG Pathways Relative abundance;
Organismal Systems (0.68%); (b) Piphillin; KEGG Pathways Relative abundance; 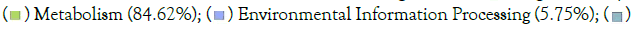 Human Diseases (5.01%);
Human Diseases (5.01%); 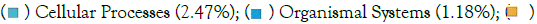 Genetic Information Processing (0.97%).
Genetic Information Processing (0.97%).

Figure 2: Extended error bar chart representation of the significant predictive functionalities as inferred by both PICRUSt2 and Piphillin. (a)
Overall, metabolism is significantly higher in PICRUSt2 analysis as compared to that of Piphillin, however, (b) a shared difference was observed
at the super-pathway level. Significance (q-value>0.05) was calculated using White’s non-parametric test with Benjamini-Hochberg FDR (false discovery rate) in STAMP. Note:  Piphillin.
Piphillin.

Figure 3: An overall comparison of the significant metabolic pathways as inferred by PICRUSt2 and Piphillin depicting a significant number
of functional features predicted by these two pipelines. Significance (q-value>0.05) was calculated using White’s non-parametric test with Benjamini-Hochberg FDR (false discovery rate) in STAMP. Note: Piphillin.
Piphillin.
Non-parametric correlation of bacteria with predictive functionality
Non-parametric Spearman’s correlation analysis resulted in a complex bacterial-functions interaction. Lactococcus showed a significant negative correlation with glycerolipid metabolism and ubiquinone and other terpenoid-quinone biosynthesis. Lactobacillus showed significant negative correlation with tryptophan metabolism, galactose metabolism, and lipoic acid metabolism while it was observed to be positively significantly correlated with sulphur metabolism. On the other hand, valine, leucine and isoleucine degradation, arginine biosynthesis and ubiquinone and other terpenoid-quinone biosynthesis was positively correlated with Leuconostoc, and negatively correlated with galactose metabolism. Furthermore, a significant negative correlation was observed between Acetobacter with pathways-tryptophan metabolism, valine, leucine and isoleucine biosynthesis, and lipoic acid metabolism. Gluconobacter also showed a significant negative correlation with phenylalanine metabolism, pentose and glucuronate interconversions, fructose and mannose metabolism, and nitrogen metabolism. Glycerolipid metabolism and ubiquinone and other terpenoid-quinone biosynthesis showed significant positive correlation with Staphylococcus, which significantly negatively correlated with propanoate metabolism. Pseudomonas showed significant negative correlation with fructose and mannose metabolism and significant positive correlation with tyrosine metabolism, valine, leucine and isoleucine degradation, arginine and proline metabolism, galactose metabolism, ubiquinone and other terpenoid-quinone biosynthesis and glutathione metabolism. Additionally, a significant positive correlation was observed between Acinetobacter with phenylalanine metabolism, streptomycin biosynthesis, ascorbate and aldarate metabolism, propanoate metabolism, nitrogen metabolism, and biosynthesis of ansamycins (Figure 4).

Figure 4: Non-parametric Spearman’s correlation of the ASV-associated predominant bacterial genera of the NFM products with a consolidated
functional feature as inferred by both PICRUSt2 and Piphillin. Here, calculation was carried out using Statistical Package for the Social Sciences
(SPSS) v20 and heatmap was generated using ClustVis. All significant correlation pairs are denoted by * (*<0.05 and **<0.01). LAB-lactic acid bacteria; AAB-acetic acid bacteria. Note:
Discussion
In this study, microbial predictive gene functional analysis from targetted-16S rRNA gene was explored using PICRUSt2 and Piphillin pipelines. Inference of predictive functionality using these two said pipelines showed a high metabolism rate, since most of these products are consortia of many metabolically active microbiota [3]. These findings are similar to recent studies reported from fermented dairy products [9-11]. The association of various metabolic pathways such as amino acid metabolism, carbohydrate metabolism, energy metabolism, lipid metabolism, metabolism of cofactors and vitamins, and other secondary metabolites with the bacterial genera indicated an active interaction of bacteria- function complexity. LAB are predominant microbiota in many ethnic fermented milk products of India followed by few AAB [28-32] Spearman’s correlation of the predominant bacterial genera with the predictive functionality resulted in a complex microbial- functions interaction in NFM products of Sikkim and Arunachal Pradesh. Metabolic activity such as amino acid metabolism is important in dairy products as they contribute to development of flavour [33]. Similarly, carbohydrate metabolism does also play a major role in flavour and aroma development in milk fermentation [34]. The abundance of functional pathways related to metabolism of amino acids, lipid, energy, and carbohydrates were earlier reported in fermented milk and milk products [35,9,36,10]. A high correlation of functional properties and LAB have also been reported in cheeses [37] since LAB are the most predominant microorganisms in fermented milk products [38,10]. We observed a positive correlation of Staphylococcus with the predictive metabolic features of these NFM products, and interestingly, Staphylococcus is metabolically active in dairy products playing functional activities such as amino acid metabolism, carbohydrate metabolism, lipid metabolism and nitrogen metabolism [39]. We also observed the presence of significant correlation of bacteria with cofactors and vitamins metabolism such as ubiquinone and other terpenoid- quinone biosynthesis and lipoic acid metabolism, which are essential for other microbial metabolism [40]. Apart from LAB, AAB have also contributed to many functional features in NFM products; AAB involve in protein metabolism, production of secondary metabolites and volatile compounds [41,42].
Functional profiles from both PICRUSt2 and Piphillin were normalized using MUSiCC [25], which is a marker gene-based method which uses universal single-copy genes for biasness correction of gene abundances [43]. Normalization using MUSiCC have proven necessary for gene functional studies [44], rescaling the abundant predicted KOs to the actual average gene copy number, correcting several known biases [45]. Piphillin is usually applied in human clinical samples (Iwai et al. 2016); whereas PICRUSt2 is widely used for environmental samples [7]. However, these pipelines have also been widely used in dairy products [11].
From our present analysis, PICRUSt2 analysis generated more predicted KOs and KEGG pathways in comparison to that of Piphillin. Though, significant differences were observed, however, there are functions which were predicted only from PICRUSt2 and missing in Piphillin and vice versa. Therefore, consolidated predictive functions from both these pipelines are necessary for a comprehensive outlook into the potential of bacteria associated with NFM products. Though predictive functionality study of the microbiota associated with NFM products at present is only speculations using bioinformatics tools, a general outlook into the potentiality of functions may be studied and compared. Nonetheless, in the absence of shotgun metagenomics data, using PICRUSt2 and Piphillin serves to be the reliable analysis for microbial predictive gene function.
Conclusion
Bacterial community in NFM products showed many functional features with many important health benefits to consumers. We applied PICRUSt2 and Piphillin tools to infer the predictive functional features of microbiota associated with the ethnic fermented milk products of India. The use of bioinformatics tools is cost-effective and straightforward where potential functional features can be mined using only structural gene, in this case, 16S rRNA gene, in the absence of shotgun metagenomics. Therefore, such studies may be used for future comparison with detailed gene functionality studies of other fermented foods elsewhere.
Acknowledgements
The authors are grateful to the Department of Biotechnology, Govt. of India through the DAICENTER project. HNJS is grateful to DBT for Junior Research Fellowship.
Funding
This current research is supported by Department of Biotechnology, Govt. of India through DBT-AIST International Centre for Translational and Environmental Research (DAICENTER) project.
Authors' Contributions
HNJS did analysis and bioinformatics analysis. JPT has supervised the bioinformatics analysis and finalised the manuscript.
Availability of Data and Materials
Raw sequences were accessed from MG-RAST server having the MG-RAST ID number 4732361 to 4732414. The same were accessed from NCBI database server under the BioProject No. PRJNA661385 with accession numbers SAMN16056817 to SAMN16056870.
Declaration of Competing Interest
The authors declare that they have no competing interests.
REFERENCES
- Rai R, Shangpliang HN, Tamang JP. Naturally fermented milk products of the Eastern Himalayas. J Ethnic Foods. 2016; 3(4):270-275.
[Crossref], [Google Scholar]
- Tamang JP, Jeyaram K, Rai AK, Mukherjee PK. Diversity of beneficial microorganisms and their functionalities in community-specific ethnic fermented foods of the Eastern Himalayas. Food Res Inter. 2021; 148:110633.
[Crossref], [Google Scholar], [PubMed]
- Shangpliang HN, Rai R, Keisam S, Jeyaram K, Tamang JP. Bacterial community in naturally fermented milk products of Arunachal Pradesh and Sikkim of India analysed by high-throughput amplicon sequencing. Scientific Reports. 2018; 8(1):1.
[Crossref], [Google Scholar], [PubMed]
- Quince C, Walker AW, Simpson JT, Loman NJ, Segata N. Shotgun metagenomics, from sampling to analysis. Nat Biotechnol. 2017; 35(9):833-844.
[Crossref], [Google Scholar], [PubMed]
- Ortiz‐Estrada ÁM, Gollas‐Galván T, Martínez Córdova LR, Martínez‐Porchas M. Predictive functional profiles using metagenomic 16S rRNA data: A novel approach to understanding the microbial ecology of aquaculture systems. Rev Aquac. 2019; 11(1):234-245.
[Crossref], [Google Scholar]
- Bokulich NA, Ziemski M, Robeson M, Kaehler B. Measuring the microbiome: Best practices for developing and benchmarking microbiomics methods. Comput Struct Biotechnol J. 2020.
[Crossref], [Google Scholar], [PubMed]
- Douglas GM, Maffei VJ, Zaneveld JR, Yurgel SN, Brown JR, Taylor CM, et al. PICRUSt2 for prediction of metagenome functions. Nat Biotechnol. 2020; 38(6):685-688.
[Crossref], [Google Scholar], [PubMed]
- Narayan NR, Weinmaier T, Laserna-Mendieta EJ, Claesson MJ, Shanahan F, Dabbagh K, et al. Piphillin predicts metagenomic composition and dynamics from DADA2-corrected 16S rDNA sequences. BMC Genomics. 2020; 21(1):1-2.
[Crossref], [Google Scholar], [PubMed]
- Zhu Y, Cao Y, Yang M, Wen P, Cao L, Ma J, et al. Bacterial diversity and community in Qula from the Qinghai–Tibetan Plateau in China. PeerJ. 2018; 6:e6044.
[Crossref], [Google Scholar], [PubMed]
- Chen X, Zheng R, Liu R, Li L. Goat milk fermented by lactic acid bacteria modulates small intestinal microbiota and immune responses. J Funct Foods. 2020; 65:103744.
[Crossref], [Google Scholar]
- Choi J, Lee SI, Rackerby B, Goddik L, Frojen R, Ha SD, et al. Microbial communities of a variety of cheeses and comparison between core and rind region of cheeses. J Dairy Sci. 2020; 103(5):4026-4042.
[Crossref], [Google Scholar], [PubMed]
- Bengoa AA, Iraporda C, Garrote GL, Abraham AG. Kefir micro organisms: Their role in grain assembly and health properties of fermented milk. J Appl Microbiol. 2019; 126(3):686-700.
[Crossref], [Google Scholar], [PubMed]
- Tamang JP, Cotter PD, Endo A, Han NS, Kort R, Liu SQ, et al. Fermented foods in a global age: East meets West. Compr Rev Food Sci Food Saf. 2020; 19(1):184-217.
[Crossref], [Google Scholar], [PubMed]
- Garcia-Burgos M, Moreno-Fernandez J, Alferez MJ, Diaz-Castro J, Lopez-Aliaga I. New perspectives in fermented dairy products and their health relevance. J Funct Foods. 2020; 72:104059.
[Crossref], [Google Scholar]
- Bolyen E, Rideout JR, Dillon MR, Bokulich NA, Abnet CC, Al-Ghalith GA, et al. Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nat Biotechnol. 2019; 37(8):852-857.
[Crossref], [Google Scholar], [PubMed]
- Callahan BJ, McMurdie PJ, Rosen MJ, Han AW, Johnson AJ, Holmes SP. DADA2: high-resolution sample inference from Illumina amplicon data. Nature Methods. 2016; 13(7):581-583.
[Crossref], [Google Scholar], [PubMed]
- Quast C, Pruesse E, Yilmaz P, Gerken J, Schweer T, Yarza P, et al. The SILVA ribosomal RNA gene database project: Improved data processing and web-based tools. Nucleic acids research. 2012; 41(D1):D590-D596.
[Crossref], [Google Scholar], [PubMed]
- Rognes T, Flouri T, Nichols B, Quince C. Mah e, F. VSEARCH: A versatile open source tool for metagenomics. PeerJ. 2016; 4:e2584.
[Crossref], [Google Scholar], [PubMed]
- Barbera P, Kozlov AM, Czech L, Morel B, Darriba D, Flouri T, et al. EPA-ng: Massively parallel evolutionary placement of genetic sequences. Sys Biol. 2019; 68(2):365-369.
[Crossref], [Google Scholar], [PubMed]
- Czech L, Stamatakis A. Scalable methods for analyzing and visualizing phylogenetic placement of metagenomic samples. PLoS One. 2019; 14(5):e0217050.
[Crossref], [Google Scholar], [PubMed]
- Louca S, Doebeli M. Efficient comparative phylogenetics on large trees. Bioinformat. 2018; 34(6):1053-1055.
[Crossref], [Google Scholar], [PubMed]
- Ye Y, Doak TG. A parsimony approach to biological pathway reconstruction/inference for genomes and metagenomes. PLoS Computational Biol. 2009; 5(8):e1000465.
[Crossref], [Google Scholar], [PubMed]
- Kanehisa M, Goto S, Sato Y, Furumichi M, Tanabe M. KEGG for integration and interpretation of large-scale molecular data sets. Nucleic Acids Res. 2012; 40(D1):D109-D114.
[Crossref], [Google Scholar], [PubMed]
- Iwai S, Weinmaier T, Schmidt BL, Albertson DG, Poloso NJ, Dabbagh K, et al. Piphillin: improved prediction of metagenomic content by direct inference from human microbiomes. PloS one. 2016; 11(11):e0166104.
[Crossref], [Google Scholar], [PubMed]
- Manor O, Borenstein E. MUSiCC: A marker gene based framework for metagenomic normalization and accurate profiling of gene abundances in the microbiome. Genome Biol. 2015; 16(1):1-20.
[Crossref], [Google Scholar], [PubMed]
- Parks DH, Tyson GW, Hugenholtz P, Beiko RG. STAMP: Statistical analysis of taxonomic and functional profiles. Bioinformatics. 2014; 30(21):3123-3124.
[Crossref], [Google Scholar], [PubMed]
- Metsalu T, Vilo J. ClustVis: A web tool for visualizing clustering of multivariate data using principal component analysis and heatmap. Nucleic Acids Res. 2015; 43(W1):W566-W570.
[Crossref], [Google Scholar], [PubMed]
- Tamang JP, Dewan S, Thapa S, Olasupo NA, Schillinger U, Wijaya A, et al. Identification and enzymatic profiles of the predominant lactic acid bacteria isolated from soft‐variety Chhurpi, a traditional cheese typical of the Sikkim Himalayas. Food Biotechnol. 2000; 14(1-2):99-112.
[Crossref], [Google Scholar]
- Dewan S, Tamang JP. Microbial and analytical characterization of Chhu-A traditional fermented milk product of the Sikkim Himalayas. 2006; 747-752.
- Dewan S, Tamang JP. Dominant lactic acid bacteria and their technological properties isolated from the Himalayan ethnic fermented milk products. Antonie van Leeuwenhoek. 2007; 92(3):343-352.
[Crossref], [Google Scholar], [PubMed]
- Ghosh T, Beniwal A, Semwal A, Navani NK. Mechanistic insights into probiotic properties of lactic acid bacteria associated with ethnic fermented dairy products. Front Microbiol. 2019; 10:502.
[Crossref], [Google Scholar], [PubMed]
- Shangpliang HN, Tamang JP. Phenotypic and genotypic characterisation of lactic acid bacteria isolated from exotic naturally fermented milk (cow and yak) products of Arunachal Pradesh, India. Inter Dairy J. 2021; 118:105038.
[Crossref], [Google Scholar]
- Yvon M, Rijnen L. Cheese flavour formation by amino acid catabolism. Intern Dairy J. 2001; 11(4-7):185-201.
[Crossref], [Google Scholar]
- Pan DD, Wu Z, Peng T, Zeng XQ, Li H. Volatile organic compounds profile during milk fermentation by Lactobacillus pentosus and correlations between volatiles flavor and carbohydrate metabolism. J Dairy Sci. 2014; 97(2):624-631.
[Crossref], [Google Scholar], [PubMed]
- Ramezani M, Hosseini SM, Ferrocino I, Amoozegar MA, Cocolin L. Molecular investigation of bacterial communities during the manufacturing and ripening of semi-hard Iranian Liqvan cheese. Food Microbiol. 2017; 66:64-71.
[Crossref], [Google Scholar], [PubMed]
- Yasir M, Bibi F, Hashem AM, Azhar EI. Comparative metagenomics and characterization of antimicrobial resistance genes in pasteurized and homemade fermented Arabian laban. Food Res Intern. 2020; 137:109639.
[Crossref], [Google Scholar], [PubMed]
- Yang C, Zhao F, Hou Q, Wang J, Li M, Sun Z. PacBio sequencing reveals bacterial community diversity in cheeses collected from different regions. J Dairy Sci. 2020; 103(2):1238-1249.
[Crossref], [Google Scholar], [PubMed]
- Rezac S, Kok CR, Heermann M, Hutkins R. Fermented foods as a dietary source of live organisms. Front Microbiol. 2018; 9:1785.
[Crossref], [Google Scholar], [PubMed]
- Leroy S, Even S, Micheau P, de La Foye A, Laroute V, Le Loir Y, et al. Transcriptomic analysis of staphylococcus xylosus in solid dairy matrix reveals an aerobic lifestyle adapted to rind. Microorganis. 2020; 8(11):1807.
[Crossref], [Google Scholar], [PubMed]
- Yao Y, Zhou X, Hadiatullah H, Zhang J, Zhao G. Determination of microbial diversities and aroma characteristics of Beitang shrimp paste. Food Chem. 2021; 344:128695.
[Crossref], [Google Scholar], [PubMed]
- Illeghems K, Weckx S, de Vuyst L. Applying meta-pathway analyses through metagenomics to identify the functional properties of the major bacterial communities of a single spontaneous cocoa bean fermentation process sample. Food Microbiol. 2015; 50:54-63.
[Crossref], [Google Scholar], [PubMed]
- Ai M, Qiu X, Huang J, Wu C, Jin Y, Zhou R. Characterizing the microbial diversity and major metabolites of Sichuan bran vinegar augmented by Monascus purpureus. Int J Food Microbiol. 2019; 292:83-90.
[Crossref], [Google Scholar], [PubMed]
- Noecker C, McNally CP, Eng A, Borenstein E. High-resolution characterization of the human microbiome. Transl Res. 2017; 179:7-23.
[Crossref], [Google Scholar], [PubMed]
- Vincent AT, Derome N, Boyle B, Culley AI, Charette SJ. Next-generation Sequencing (NGS) in the microbiological world: How to make the most of your money. J Microbiol Methods. 2017; 138:60-71.
[Crossref], [Google Scholar], [PubMed]
- Manor O, Borenstein E. Systematic characterization and analysis of the taxonomic drivers of functional shifts in the human microbiome. Cell Host Microbe. 2017; 21(2):254-267.
[Crossref], [Google Scholar], [PubMed]
Citation: Shangpliang HNJ, Tamang JP (2022) Predictive Functionality of Bacteria in Naturally Fermented Milk Products of India Using PICRUSt2 and Piphillin Pipelines. J Data Mining Genomics Proteomics. 13:244.
Copyright: © 2022 Shangpliang HNJ, et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.