


Indexed In
- Academic Journals Database
- Open J Gate
- Genamics JournalSeek
- Academic Keys
- JournalTOCs
- China National Knowledge Infrastructure (CNKI)
- CiteFactor
- Scimago
- Ulrich's Periodicals Directory
- Electronic Journals Library
- RefSeek
- Hamdard University
- EBSCO A-Z
- OCLC- WorldCat
- SWB online catalog
- Virtual Library of Biology (vifabio)
- Publons
- MIAR
- University Grants Commission
- Geneva Foundation for Medical Education and Research
- Euro Pub
- Google Scholar
Useful Links
Share This Page
Journal Flyer

Open Access Journals
- Agri and Aquaculture
- Biochemistry
- Bioinformatics & Systems Biology
- Business & Management
- Chemistry
- Clinical Sciences
- Engineering
- Food & Nutrition
- General Science
- Genetics & Molecular Biology
- Immunology & Microbiology
- Medical Sciences
- Neuroscience & Psychology
- Nursing & Health Care
- Pharmaceutical Sciences
Research Article - (2024) Volume 16, Issue 5
Pharmacokinetic Equivalence Study of the Oral Direct Inhibitor of Factor Xa Anticoagulant, Rivaroxaban Film-Coated Tablets, in Healthy Subjects Under Fasting and Fed Conditions
Raymond R. Tjandrawinata1,2*, Danang Agung Yunaidi3, Yantirta Indra Kurniawan3, Clarasintha Nindyatami1, Ismail Dwi Saputro3, Ima Aisyah Rahma3, Vicky Achmad Ginanjar3 and Liana W. Susanto12Department of Biotechnology, Atma Jaya Catholic University of Indonesia, Banten, Indonesia
3PT. Equilab International Bioavailability and Bioequivalence Laboratory, Jakarta, Indonesia
Received: 02-Sep-2024, Manuscript No. JBB-24-26854; Editor assigned: 04-Sep-2024, Pre QC No. JBB-24-26854 (PQ); Reviewed: 18-Sep-2024, QC No. JBB-24-26854; Revised: 25-Sep-2024, Manuscript No. JBB-24-26854 (R); Published: 02-Oct-2024, DOI: 10.35248/0975-0851.24.16.595
Abstract
Purpose: To evaluate the bioequivalence of two strengths of Rivaroxaban 10 mg and 20 mg film-coated tablets manufactured by PT Dexa Medica with the reference drug, Xarelto® film-coated tablet, manufactured by Bayer AG, Germany, imported by PT Bayer Indonesia, Indonesia.
Materials and Methods: Two studies were conducted separately under fasting condition for the 10 mg strength and fed condition for the 20 mg. The studies were open-label, randomized, single-dose, two-period, two-sequence, two-way crossover studies, which included 28 healthy adult male and female subjects in each study. The washout period was five days and 11 days for the 10 mg strength and 20 mg, respectively. The 10 mg study was completed by 28 subjects, whereas the 20 mg strength study was completed by 26 subjects. The plasma concentrations of Rivaroxaban were determined using validated Ultra-Performance Liquid Chromatography with Tandem Mass Spectrometry Detection (UPLC-MS/MS). The pharmacokinetic parameters assessed were Area Under the Curve (AUC0–t), Area Under The Curve (AUC0–∞), Maximum Concentration (Cmax), Time-to-Maximum (Tmax), and Half-life (t½). Bioequivalence was established in the Test/Reference Geometric Means Ratio (GMR), and the 90% Confidence Interval (CI) of GMR was between 80.00 and 125.00% with 0.05 alpha for AUC0-t and Cmax.
Results: The GMR (90% CI) of the Test/Reference formulation of Rivaroxaban 10 mg was 91.51% (82.41%- 101.63%) for Cmax and 98.32% (92.20%-104.83%) for AUC0–t under fasting condition. The GMR (90% CI) of the Test/Reference formulation of Rivaroxaban 20 mg was 92.76% (87.60%-98.22%) for Cmax and 94.35% (90.14%- 98.76%) for AUC0–t under fed condition. The 90% CI of the test/reference drug from both strengths was within the bioequivalence acceptable range. Adverse events were reported in both studies, two in the 10 mg strength study and one in the 20 mg strength. All cases were mild, and no serious adverse events were reported.
Conclusion: The studies indicated that Rivaroxaban 10 mg and 20 film-coated tablets were bioequivalent with the reference drugs under fasting and fed conditions. The subjects well tolerated both formulations
Keywords
Anticoagulant; Bioavailability; Bioequivalence; Rivaroxaban; Generic; Pharmacokinetics
Introduction
Rivaroxaban (C19H18ClN3O5S) was the first oral direct factor Xa inhibitor to be marketed which belongs to the new family of anticoagulants known as Direct Oral Anticoagulants (DOACs). DOACs are a kind of medication that is recently developed as fastacting oral medicine. Due to the fact that DOACs do not require the heparin bridging phase, they have mostly replaced Vitamin K Antagonist (VKAs) since their launch [1,2]. Chemically, Rivaroxaban is defined as 5-chloro-N-(((5S)-2-oxo-3-(4-(3-oxomorpholin-4-yl) phenyl)-1,3-oxazolidin-5-yl)methyl)thiophene-2-carboxamide with the chemical structure described in Figure 1. Almost insoluble in water, Rivaroxaban is a small molecule with a molecular weight of 435.9 g/mol that binds to plasma proteins with a high proportion (92%-95%) in humans with serum albumin being the primary binding component [3,4].

Figure 1: Chemical structure of Rivaroxaban.
Rivaroxaban is currently approved in several nations, including Europe, the United States of America, and other areas, to prevent Venous Thromboembolism (VTE) in patients having total hip or knee replacement surgery [5,6]. Under stable conditions, Rivaroxaban's inhibition of factor Xa can lower or reduce the production of thrombin. The prothrombinase complex's active component, factor Xa, catalyzes the transformation of prothrombin (factor II) into thrombin (factor IIa). This inhibits the process of spreading coagulation by blocking factor Xa produced through both intrinsic and extrinsic coagulation pathways [4]. The medication is being tested widely in several clinical contexts, such as the prevention of cardiovascular events in patients with acute coronary syndrome, the prevention of stroke in those with atrial fibrillation, and hospitalized medically ill patients and VTE prevention [7-9].
Regardless of age, weight, or gender, Rivaroxaban has been shown to have a very predictable and consistent clinical pharmacology profile across all patient populations studied, which supports the use of a fixed-dose regimen without the need for dose adjustments. This effectively removes the requirement for regular coagulation monitoring [10,11]. Studies that have been published have shown that a broad variety of people with a range of thromboembolic disorders can benefit from the medication. In this way, Rivaroxaban offers greater convenience for the treatment and prevention of venous and specific arterial thrombotic disorders than the existing standards of care [12]. Throughout the initial phases of clinical assessment, the pharmacokinetic profile of Rivaroxaban in healthy individuals demonstrated a positive safety and tolerability profile. Within 2-4 h of oral administration, Rivaroxaban reaches its peak plasma concentration (Cmax) due to its quick absorption and high bioavailability (80%-100%) in a 10 mg dosage. A meal does not influence the area under the plasma concentration time curve (AUC) or Cmax of Rivaroxaban; thus, it is safe to take the 10 mg tablet with or without meals. The oral absorption of the medication is nearly complete for the 10 mg tablet. Good bioavailability can be obtained by taking Rivaroxaban at higher dosages (15 mg and 20 mg) with meals. After several doses in healthy subjects, no meaningful accumulation was seen at any level beyond the steady state. Rivaroxaban is eliminated from plasma with a 7-11 h terminal half-life on average. The inter-individual variability (Coefficient Variation (CV)) of Rivaroxaban's pharmacokinetics ranges between 30% and 40%, indicating a certain amount of variability [11,13,14].
The objective of this study was to assess if the Rivaroxaban 10 mg film-coated tablet formulation by PT Dexa Medica is equivalent to that of the comparator drug (Xarelto® 10 mg film-coated tablet, produced by Bayer AG, Germany, imported by PT Bayer Indonesia, Indonesia) when given to healthy subjects under fasting conditions. Additionally, since food affecting the bioavailability of the drug at the higher dose, we also evaluated through the study whether under fed condition the bioavailability of PT Dexa Medica’s formulation of Rivaroxaban 20 mg film-coated tablet is equivalent to that of the comparator drug (Xarelto® 20 mg film-coated tablet, produced by Bayer AG, Germany, imported by PT Bayer Indonesia, Indonesia). A drug is considered bioequivalent if its Cmax and AUC are 80%- 125% of those of the reference drug [15,16].
Materials and Methods
Study subjects and design
Two studies were conducted separately under fasting condition for the 10 mg strength and fed condition for the 20 mg Rivaroxaban. The studies were open-label, randomized, single-dose, two-period, two-sequence, two-way crossover studies, which included 28 healthy adult male and female subjects in each study. The washout duration of the 10 mg (fasting) and 20 mg (fed) study was 7 and 11 days, respectively. The studies were performed according to the guideline on the Investigation of Bioequivalence, EMA, London, 2010; ASEAN Guideline for the Conduct of Bioequivalence Study, Lao PDR, 2015; and Indonesian guidelines, Tata Laksana Uji Bioekivalensi, Badan Pengawas Obat dan Makanan (BPOM), Jakarta, 2022.
The study protocol was registered in ClinicalTrials.gov with the trial registry number of NCT06558058 and NCT06558045 for the 10 mg and 20 mg strength, respectively. Before starting the trials, the Faculty of Medicine Ethics Committee at Universitas Indonesia approved study protocols and informed consent statements. The studies adopted the appropriate versions of the Declaration of Helsinki and the International Council for Harmonisation, GCP (EMA/CHMP/ICH/135/1995). All study subjects signed the written informed consent statements before beginning the screening evaluation process.
Both trials included healthy, non-smoking female and male adult volunteers aged 18-55 years with a Body Mass Index (BMI) ranging from 18 to 25 kg/m2. The subjects should have their Prothrombin Time (PT) and activated Partial Thromboplastin Time (aPTT) values within normal range and have normal renal function with acceptable creatinine clearance (CrCl) >50 ml/min. The study participants' PT and aPTT readings should be within the normal range, as should their renal function, with a CrCl of at least 50 ml/ min. The patients showed no signs of severe illness or clinically significant abnormal laboratory values during screening.
Subjects who had a history of allergy or hypersensitivity or contraindication to Rivaroxaban or factor Xa inhibitors or allied drugs; pregnant or lactating females; subjects with any clinically significant abnormal values during screening; significant liver disease, hematology and Electrocardiogram (ECG) abnormalities; history of anaphylaxis or angioedema, drug or alcohol abuse within 12 months prior to screening, bleeding or coagulative disorders, and significant head or spinal cord injury or recent surgery on the brain, spinal cord or eyes; any medical condition that might significantly alter the absorption, distribution, metabolism or excretion of the study drug; positive results for Hepatitis B surface antigen (HBsAg), anti-HCV, anti-HIV, or COVID-19 rapid antigen test; participated in any clinical trial within the past 90 days calculated from the last visit until this study’s first dosing day; subjects with difficulty in accessibility of veins in left or right arm; donated or had significant blood loss within 90 days before this study’s first dosing day; or intake of any prescription or nonprescription drugs, food supplements, or herbal medicines within 14 days of the first dosing day were excluded from the study.
Study products
The test drug for Rivaroxaban 10 mg film-coated tablet (batch no. K-10535-00-F-SCU-2) was manufactured by PT Dexa Medica, Indonesia. Xarelto® 10 mg film-coated tablet (batch no. BXJLZH1), manufactured by Bayer AG, Germany, imported by PT Bayer Indonesia, Indonesia, served as the reference drug for the 10 mg strength study.
Rivaroxaban 20 mg film-coated tablet (batch no. K-10537-00-FSCU- 2) manufactured by PT Dexa Medica-Indonesia was used as the test drug for the 20 mg study. The reference drug used was Xarelto® 20 mg film-coated tablet (batch no. BXJLBJ1) manufactured by Bayer AG, Germany, imported by PT Bayer Indonesia, Indonesia.
Each subject in the crossover studies received one Test drug (T) and one Reference drug (R) by a random sequence of TR or RT determined by block randomization and Table of Random Numbers from Dixon & Massey, 1983 [17]. Both studies used an open-label design. Nevertheless, the randomization code was not accessible to the bioanalytical and statistical analysis department.
Treatment phase and blood sampling
Eligible subjects were scheduled to arrive a day before the drug dosing at PT Equilab. In the Rivaroxaban 10 mg film-coated tablets fasting study, subjects were requested to fast, except for mineral water, for 8 h prior to the drug administration. Day-1 began with collecting a 5 ml pre-dose pharmacokinetic blood sample on the morning of the dosing day. One T or one R was dosed to each subject along with 200 ml of water. Five ml of blood were extracted by venipuncture using a 22-gauge needle at intervals of 0.33, 0.67, 1.00, 1.33, 1.67, 2.00, 2.33, 2.67, 3.00, 3.33, 3.67, 4.00, 4.33, 4.67, 5.00, 6.00, 8.00, 12.00, 24.00 and 36.00 h following the administration of the drug at each period. Regarding the fed study of Rivaroxaban 20 mg film-coated tablets, the pre-dose blood sample was taken on Day-1 before the individuals had a high-fat breakfast that they were expected to consume within 30 min After breakfast, 200 ml of water was given with the study drug. After the drug was administered, at 0.50, 1.00, 1.33, 1.67, 2.00, 2.33, 2.67, 3.00, 3.33, 3.67, 4.00, 4.33, 4.67, 5.00, 5.50, 6.00, 8.00, 12.00, 24.00, and 36.00 h, post-dose blood samples were taken for the fed study.
The pre-dose and post-dose blood samples were collected in vacuum polyethylene tubes containing Ethylenediaminetetraacetic Acid Tripotassium Salt Dehydrate (K3EDTA). Blood samples were centrifuged at 1538 ± 10 g for 15 min at room temperature to separate the plasma. All plasma samples were kept in a freezer at -200C ± 50C until assayed.
The subjects' vital signs, including blood pressure, pulse rate, respiratory rate, and body temperature, and adverse events were observed at 2.00, 4.00, 6.00, 8.00, 12.00, 24.00, and 36.00 h postdose.
Fluid intake was restricted from 1 h pre-dose to 2 h post-dose, except for the water provided during dosing in the fasting study. Subjects of fasting and fed studies were prohibited from consuming xanthine-containing food or beverages, fruit juices, and alcoholbased products for 24 h before and during the entire sampling days. Fruit juices, items containing alcohol, and foods or beverages containing xanthine were off-limits to study subjects for 24 h before and throughout the sample days for both fed and fasted studies. During the trial and for 21 days before the initial dosing day, concomitant drug use was prohibited. If any concomitant medicine is required during the trial, it must be documented in the Electronic Case Report Form (e-CRF). For the 10 mg fasting study and the 20 mg fed study, the washout intervals between each dose were 7 and 11 days, respectively. In the subsequent study period, the same procedures were repeated using the alternate study drug.
Drug concentration analysis
Apixaban was added to each 300 μL plasma sample as the internal standard and protein precipitation using acetonitrile:methanol (1:1) for extraction. After being poured into a vial, the supernatant was diluted with water. Subsequently, the aliquot underwent analysis UPLC-MS/MS utilizing an MS/MS detector Waters XEVO TQD (ANL – Xevo 1) and an LC Waters ACQUITY UPLC H-Class. The C18 Poroshell (2.7 μm, 2.1 × 50 mm) analytical column was used, with a mobile phase of acetonitrile:ammonium formate 5 mM (40:60) at a 0.4 ml/min flow rate. A fully validated UPLC-MS/MS system with sufficient sensitivity, selectivity, specificity, linearity, accuracy and precision (both within and between days) was used to measure the plasma concentrations of Rivaroxaban.
Table 1 displays the sample stability and validation results. The Lower Limit of Quantitation (LLOQ) was established at 2.01 ng/ml. All chromatograms were automatically processed using the same MassLynx software, including peak-to-peak amplitude, identification, integration, and smoothing.
| Parameters | Concentration | ||||
|---|---|---|---|---|---|
| LLOQ (2.01 ng/ml) | Low (6.03 ng/ml) | Medium (241.34 ng/ml) | High (603.35 ng/ml) | ULOQ (804.47 ng/ml) | |
| Precisiona | |||||
| Intra-assay CV | 6.70%-16.69% | 6.05%-9.29% | 3.48%-8.64% | 2.27%-6.02% | 1.19%-3.66% |
| Inter-assay CV | 9.64% | 9.07% | 5.18% | 4.94% | 3.32% |
| Accuracya | |||||
| Intra-assay CV | -8.26%-9.45% | -10.78%-3.00% | -9.92%-1.07% | -1.88%-1.11% | 0.21%-1.11% |
| Inter-assay CV | -4.44% | -4.75% | -7.76% | -1.74% | 0.97% |
| Stability of plasma samplea | |||||
| At -20oC ± 5oC (stable until 81 days) | - | -10.78%-10.65% | - | -1.72%-7.73% | - |
| At room temperature (≤30oC, stable until 24 hours) | - | -7.53%-5.17% | - | -11.07%-1.11% | - |
| Freeze and thaw (stable until 4 cycles at -20oC±5oC) | - | -10.78%-1.99% | - | -1.72%-7.73% | - |
| Linearity | |||||
| Linear relationship between the concentration and signal intensity was established (r=0.9976 for run 1, r=0.9985 for run 2, r=0.9992 for run 3) | |||||
| Selectivity | |||||
| The % interference in the retention time of analyte and internal standard in 8 lots blank plasma with K3EDTA as the anticoagulant (comprising of 6 normal, 1 hemolysis and 1 lipemic) were ranged from 0.00% to 15.18% for analyte and 0.00% to 0.02% for internal standard. | |||||
| Specificity (concomitant medication effect) | |||||
| The % interference in the retention time of analyte and internal standard in 8 lots blank plasma with K3EDTA as the anticoagulant (comprising of 6 normal, 1 hemolysis and 1 lipemic) with presence of paracetamol, and mefenamic acid as the co-administered medication were ranged from 0.00% to 5.41% for analyte and 0.00% to 0.02% internal standard. | |||||
| Range | |||||
| The range of quantification has been established from 2.01 ng/ml to 804.47 ng/ml | |||||
| Note: a Expressed by the percent difference between the measured and actual values. | |||||
Table 1: Validation summary results of the analytical method of rivaroxaban determination in plasma samples by UPLC-MS/MS with apixaban as the internal standard.
Pharmacokinetic analysis
Several pharmacokinetic parameters were determined from the Rivaroxaban plasma concentration-time data analysis. The Cmax and Tmax were obtained directly from the observed data. The area under the plasma concentration-time curve from administration to last observed quantifiable concentration at time t (AUC0-t) was calculated using the linear trapezoidal method. The following formula was used to determine the area under the plasma concentration-time curve extrapolated to infinity (AUC0-∞),
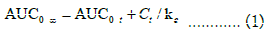
Ct was the last quantifiable concentration; ke was the terminal elimination rate constant and was determined by least-squares regression analysis during the terminal log-linear phase of the concentration-time curve. The plasma half-life (t½) was calculated according to the formula,
t½ = 0.693/ke .............................. (2)
Statistical analyses were performed using Phoenix® WinNonlin® version 8.3 (Certara L.P., St. Louis, MO, USA). After converting the data to their Logarithmic (ln) values, Analysis of Variance (ANOVA) was used to analyze AUC0-t and Cmax. The Wilcoxon matched-pairs signed-rank test was utilized to analyze the Tmax comparison between groups. Depending on the normality of the data distribution, the Student's paired t-test or the Wilcoxon matched-pairs test was used to evaluate the t½ difference. Bioequivalence was accepted if the 90% CI were within the 80.00%-125.00% range with 0.05 alpha for the test/reference GMR for AUC0-t and Cmax.
Results
Rivaroxaban 10 mg film-coated tablet (fasting condition study)
A total of 28 healthy Indonesian subjects (12 males and 16 females) aged 20 to 49 years with BMI values of 18.07-24.98 kg/m2 were enrolled in the study. All subjects completed this Rivaroxaban 10 mg film-coated tablet bioequivalence study under fasting conditions.
The mean plasma Rivaroxaban concentration-time profile in subjects (n=28) after single-dose oral administration of test and reference formulation is plotted in Figure 2. The results of the pharmacokinetic parameter (AUC0-t, AUC0-∞, Cmax, Tmax, and t½) and GMR 90% CI from the 10 mg Rivaroxaban test and reference formulation are presented in Table 2. The T/R GMR (90% CI) for Rivaroxaban 10 mg were 98.32% (92.20%-104.83%) for AUC0-t; 91.51% (82.41%–101.63%) for Cmax; and 97.97% (91.94%- 104.40%) for AUC0-∞. Under fasting conditions, the Rivaroxaban 10 mg tablets Cmax and AUC0-t GMR 90% CIs fell between 80.00 and 125.00%, the range specified by the bioequivalence criteria. The Tmax and t½ of the test and reference drug did not differ significantly (p>0.05).

Figure 2: The mean plasma-time concentration profile of Rivaroxaban (n=28) after a single oral dose of the test drug, Rivaroxaban 10 mg film-coated tablets and the reference drug, Xarelto® 10 mg film-coated tablet, under fasting condition-linear scale.
| Parameter | Rivaroxaban Dexa Medica (Test)a | Xarelto® Bayer (Reference)a | GMR T/R (90% CI)b | %CV | Power (%) |
|---|---|---|---|---|---|
| Rivaroxaban 10 mg | |||||
| Cmax (ng.ml-1)c |
219.24 (66.88) | 234.31 (68.08) | 91.51% (82.41%-101.63%) |
23.30 | 96.74 |
| AUC0-t (ng.h.ml-1)c |
1745.23 (556.35) | 1752.57 (494.71) | 98.32% (92.20%-104.83%) |
14.15 | 99.99 |
| AUC0–∞ (ng.h.ml-1)c |
1800.74 (550.62) | 1815.12 (487.15) | 97.97% (91.94%-104.40%) |
14.02 | - |
| t1/2 (h) | 7.27 (1.62) | 7.58 (2.14) | NSe | - | - |
| Tmax (h)d | (1.00-5.00) | 2.33 (0.67-4.67) |
NSf | - | - |
| Rivaroxaban 20 mg | |||||
| Cmax (ng.ml-1)c |
490.83 (71.48) | 530.09 (86.01) | 92.76% (87.60%-98.22%) |
12.10 | >99.99 |
| AUC0-t (ng.h.ml-1)c |
3869.79 (594.17) | 4134.54 (837.27) | 94.35% (90.14%-98.76%) |
9.65 | >99.99 |
| AUC0–∞ (ng.h.ml-1)c |
3954.13 (612.94) | 4219.66 (870.37) | 94.51% (90.23%-98.99%) |
9.78 | - |
| t1/2 (h) | 6.23 (1.01) | 6.34 (1.17) | NSe | - | - |
| Tmax (h)d | 4.33 (2.33-5.50) |
4.33 (2.00-5.50) |
NSf | - | - |
Note: aThe values are expressed as mean Standard Deviation (SD); bBioequivalence criterion defined as 90% CI of the GMR of the test formulation/reference drug is between 80.00% and 125.00% for AUC0–t and Cmax; cStatistical calculations for AUC0–t and Cmax were based on ln-transformation data; dThe values are expressed as median (range); eAnalysis was performed using the Student’s paired t-test; fAnalysis was performed using the Wilcoxon matched-pairs test.
Abbreviations: AUC: Area Under the plasma concentration-time Curve; AUC0–t: AUC from time zero to the last observed quantifiable concentration; AUC0–∞: AUC from time zero to infinity; Cmax: the Maximum Plasma Concentration; CI: Confidence Interval; CV: Coefficient of Variation; NS: Not Significant; R: Reference formulation; SD: Standard Deviation; T: Test formulation; t1/2: Terminal half-life; Tmax: Time to Cmax
Table 2: Pharmacokinetic parameters and statistical comparison of test and reference drug of Rivaroxaban 10 mg film-coated tablets under fasting conditions (n=28) and Rivaroxaban 20 mg film-coated tablets under fed conditions (n=26) in healthy Indonesian subjects.
Subjects receiving the test formulation had two types of adverse events; headaches (1 event) and elevated blood pressure (2 events), which occurred in 3.57% and 1.79% of dosed study subjects, respectively. The severity of each case was mild. While headaches were definitely related to the trial drug, elevated blood pressure is thought to be unlikely related.
Rivaroxaban 20 mg film-coated tablet (fed condition study)
The Rivaroxaban 20 mg film-coated tablet bioequivalence study, carried out under fed conditions, included 28 healthy Indonesian male and female participants. The participants comprised 15 males and 13 females aged 21 to 53 years, with BMI scores ranging from 18.03 to 24.91 kg/m2. One participant withdrew after blood sample during period 1, and one subject did not participate in period 2 due to personal reasons. Therefore, the pharmacokinetic characteristics and plasma levels of Rivaroxaban were determined by analyzing blood samples from 26 subjects.
The pharmacokinetic parameter values (AUC0-t, AUC0-∞, Cmax, Tmax, and t½) and the GMR 90% CIs results of the test formulation/ reference formulation of 20 mg Rivaroxaban under fed condition are displayed in Table 2. Figure 3 represents the mean plasma concentration-time profile in subjects (n=26) after the single oral dose administration of test and reference formulation of 20 mg Rivaroxaban under fed conditions. The GMR (90% CI) of the T /R for Rivaroxaban 20 mg were 94.35% (90.14%-98.76%) for AUC0-t; 92.76% (87.60%-98.22%) for Cmax and 94.51% (90.23%- 98.99%) for AUC0-∞. The 90% CIs of the Cmax and AUC0-t ratio of Rivaroxaban 20 mg tablet under fed condition fell within the bioequivalence criteria acceptance range. No significant difference was observed in the Tmax and t½ values between the test Rivaroxaban and the reference drugs (p>0.05).

Figure 3: The mean plasma-time concentration profile of Rivaroxaban (n=26) after a single oral dose of the test drug, Rivaroxaban 20 mg film-coated tablets and the reference drug, Xarelto® 20 mg film-coated tablet, under fed condition-linear scale.
During the trial, two (3.70%) adverse events resulting in elevated blood pressure were noted in the comparator group. Both side effects were deemed minor and unlikely to have been caused by the study drug.
Discussion
Under the trade name Xarelto®, Rivaroxaban was initially approved by the European Medicines Agency (EMEA) in 2008 and the United States Food and Drug Administration (USFDA) in 2011. Because of patent/exclusivity, generic versions of Rivaroxaban are not currently marketed in the United States of America; nevertheless, they are already accessible internationally. The generic formulation of Rivaroxaban produced by PT Dexa Medica-Indonesia evaluated in this study is among the first generics of Rivaroxaban developed and registered in Indonesia. A bioequivalence study is a regulatory requirement that generic medications must meet in order to be granted marketing authorization. A generic medication's bioequivalence with its reference drug implies that both will have comparable clinical effects in patients, ensuring the generic's safety and efficacy as an alternative for the reference.
The bioequivalence of two strengths of Rivaroxaban film-coated tablets (manufactured by PT Dexa Medica, Indonesia) with the reference medication (Xarelto®, manufactured by Bayer AG, Germany) was investigated in two separate single-dose, twoperiod, two-sequence, two-way crossover studies: 10 mg strength administered under fasting conditions and 20 mg strength administered under fed conditions. The rate and extent of absorption, denoted by Cmax and AUC, respectively, were used to evaluate the bioequivalence of the Test drug (T) and the Reference drug (R) [18]. Bioequivalence is concluded when the 90% CI for the ratio pharmacokinetic parameters Cmax and AUC0-t from the test drug with the reference meets the bioequivalence acceptance interval of 80.00%-125.00% [19].
Both studies enrolled 28 healthy subjects each. The enrolled subjects completed the 10 mg study, while 26 subjects completed the 20 mg study. The number of subjects who completed the studies was considered sufficient as it ensured adequate power to confirm the statistical conclusion presented in (Table 2).
The GMR (90% CI) of AUC0-t and Cmax findings for both the 10 mg strength under fasting state and the 20 mg fed state were within the acceptance range for bioequivalence. The findings demonstrated that the reference medication, Xarelto®, and the Rivaroxaban 10 mg and 20 mg produced by PT Dexa Medica, Indonesia, are biopharmaceutically equivalent. The bioequivalence studies further bridge the preclinical studies and clinical trials of the reference drugs to be applicable for the generic products [19]. By confirming the bioequivalence between both products, the generic versions of Rivaroxaban are interchangeable with the reference in clinical settings.
The different study designs for each strength aligned with the European Medicines Agency (EMA) Rivaroxaban film-coated tablets' product-specific bioequivalence guidance. Based on this guidance, a fed study should be carried out for the higher strengths (15 mg and 20 mg) and a fasting study for the lower strengths (2.5 mg and 10 mg) because the effects of food vary depending on the strength [20]. When fasting, Rivaroxaban has an absolute bioavailability that is dose-dependent, where its bioavailability decreases at higher doses. For both the 2.5 mg and 10 mg doses, the estimated absolute bioavailability ranged from 80% to 100% and was unaffected by meals. When fasting, the 20 mg strength has a decreased absolute bioavailability of approximately 66%, which is greatly increased by food consumption. It has been observed that food increases the 20 mg strength's bioavailability by 39% for AUC and 76% for Cmax. Therefore, to ensure optimal drug exposure, the Rivaroxaban prescribing label recommends taking the higher strengths 15 mg and 20 mg with meals, whereas the lower strengths can be taken with or without food [21].
Rivaroxaban’s Cmax occurs 2-4 h after the medication is administered [21]. These results align with those of our study investigation, wherein the tested generic oral formulation containing 10 mg of Rivaroxaban reached Cmax by 2.67 (1.00-5.00) h during fasting condition and for the 20 mg strength by 4.33 (2.33-5.50) h within the fed state. The results were not significantly different from the references used in the studies. Prior studies on the 10 mg strength when fasting and the 20 mg strength when fed found comparable Tmax values [22,23]. The half-life of both 10 mg and 20 mg strengths were 7.27 (1.62) h and 6.23 (1.01) h, respectively. The half-life results aligned with the reported terminal half-life value of Rivaroxaban in healthy adults aged 20-45 years, which is 5-9 h [21]. While the results of our investigation and a prior study's t1/2 for the 10 mg strength were similar [22]. Other studies revealed a higher half-life value [23,24]. Our study results showed that the T were not significantly different from the reference product, further supporting that the test and reference products had comparable rates of elimination.
As a non-vitamin K oral anticoagulant, Rivaroxaban is associated with increased risk of bleeding [21,25]. Generally, Rivaroxabantreated participants' mortality and bleeding risk increased with age, renal impairment, and diabetes [26]. No bleeding was observed during the conduct of our studies. The adverse events encountered during the studies were mild and well tolerated by the subjects.
Conclusion
The studies concluded that Rivaroxaban film-coated tablets 10 mg and 20 mg (manufactured by PT Dexa Medica) were bioequivalent to the reference drug, Xarelto® film-coated tablet (manufactured by Bayer AG, Germany, imported by PT Bayer Indonesia) in healthy Indonesian subjects under fasting and fed condition, respectively. This study results safely confirm that both strengths of the test formulations are interchangeable with the reference formulations in clinical practice. Adverse events reported in the studies were mild and well tolerated by the study subjects.
Conflict of Interest
The authors declare that there is no conflict of interest regarding the publication of this paper.
Funding Statement
The authors declare that PT Dexa Medica, Indonesia, completely funded the studies. PT Equilab International, Indonesia, an internationally recognized Bioequivalence Laboratory, developed the study protocol and design and carried out all technical, clinical, and analytical aspects of the bioequivalence studies independently, without interference from the study sponsor. RRT, LWS, and CN are PT Dexa Medica employees who were responsible for drafting and reviewing the study manuscript.
Acknowledgment
We greatly appreciate all the volunteers for their participation and cooperation during the study.
References
- Barnes GD, Mouland E. Peri-procedural management of oral anticoagulants in the DOAC era. Prog Cardiovasc Dis. 2018;60(6):600-606.
[Crossref] [Google Scholar] [PubMed]
- Oyakawa T, Fukumitsu M, Ebihara A, Shiga T. Relevance of non-bridging therapy with heparin during temporary interruption of direct oral anticoagulants in patients with cancer-associated venous thromboembolism. Ann Vasc Dis. 2022;15(2):121-125.
[Crossref] [Google Scholar] [PubMed]
- Sweetman SC. Martindale The Complete Drug Reference 36th Edition. Pharmaceutical Press. 2009:1389.
- Perzboccccrn E, Roehrig S, Straub A, Kubitza D, Mueck W, Laux V. Rivaroxaban : A new oral factor Xa inhibitor. Arterioscler Thromb Vasc Biol. 2010;30(3):376-381.
[Crossref] [Google Scholar] [PubMed]
- Loganathan V, Hua A, Patel S, Gibbons C, Vizcaychipi MP. Efficacy and safety of Rivaroxaban thromboprophylaxis after arthroplasty of the hip or knee: Retrospective cohort study. Ann R Coll Surg Engl. 2016;98(7):507-515.
[Crossref] [Google Scholar] [PubMed]
- Anderson DR, Dunbar M, Murnaghan J, Kahn SR, Gross P, Forsythe M, et al. Aspirin or Rivaroxaban for VTE prophylaxis after hip or knee arthroplasty. N Engl J Med. 2018;378(8):699-707.
[Crossref] [Google Scholar] [PubMed]
- Raskob GE, Ageno W, Albers G, Elliott CG, Halperin J, Maynard G, et al. Benefit–risk assessment of rivaroxaban for extended thromboprophylaxis after hospitalization for medical illness. J Am Heart Assoc. 2022;11(20):026229.
[Crossref] [Google Scholar] [PubMed]
- Mega JL, Braunwald E, Wiviott SD, Bassand JP, Bhatt DL, Bode C, et al. Rivaroxaban in patients with a recent acute coronary syndrome. N Engl J Med. 2012;366(1):9-19.
[Crossref] [Google Scholar] [PubMed]
- Alberts M, Chen YW, Lin JH, Kogan E, Twyman K, Milentijevic D. Risks of stroke and mortality in atrial fibrillation patients treated with Rivaroxaban and warfarin. Stroke. 2020;51(2):549-555.
[Crossref] [Google Scholar] [PubMed]
- Xu XS, Moore K, Burton P, Stuyckens K, Mueck W, Rossenu S, et al. Population pharmacokinetics and pharmacodynamics of Rivaroxaban in patients with acute coronary syndromes. Br J Clin Pharmacol. 2012;74(1):86-97.
[Crossref] [Google Scholar] [PubMed]
- Imberti D, Benedetti R. Practical management of Rivaroxaban for the treatment of venous thromboembolism. Clin Appl Thromb Hemost. 2015;21(4):309-318.
[Crossref] [Google Scholar] [PubMed]
- Bratsos S. Pharmacokinetic properties of Rivaroxaban in healthy human subjects. Cureus. 2019 Aug;11(8):1-6.
[Crossref] [Google Scholar] [PubMed]
- Mueck W, Lensing AW, Agnelli G, Decousus H, Prandoni P, Misselwitz F. Rivaroxaban: Population pharmacokinetic analyses in patients treated for acute deep-vein thrombosis and exposure simulations in patients with atrial fibrillation treated for stroke prevention. Clin Pharmacokinet. 2011;50:675-686.
[Crossref] [Google Scholar] [PubMed]
- Xarelto® Summary of Product Characteristics. Pharma BS. 2023.
- Dan Pk, Indonesia Mr, Bioekivalensi Pu. Badan Pengawas Obat dan Makanan. 2004.
- Guidance on the Investigation of Bioavailability and Bioequivalence. The European Agency for the Evaluation of Medicinal Products (EMA), Committee for Proprietary Medicinal Products (CPMP). CPMP/EWP/QWP/1401/98 Rev1. 2010.
- Dixon WJ, Massey Jr FJ. Introduction to statistical analysis. 1983.
- Bioequivalence Studies with Pharmacokinetic Endpoints for Drugs Submitted under an Abbreviated New Drug Applications (ANDA). USFDA, Center for Drug Evaluation and Research (CDER). 2021.
- European Medicines Agency. Guideline on the investigation of bioequivalence. Committee for Medicinal Products for Human Use (CHMP). 2010.
- Peri-procedural management of oral anticoagulants in the DOAC era
- Janssen Pharmaceuticals. Xarelto (Rivaroxaban) Prescribing Information. 2023.
- Ding S, Wang L, Xie L, Zhou S, Chen J, Zhao Y, et al. Bioequivalence study of 2 formulations of Rivaroxaban, a narrowâ?therapeuticâ?index drug, in healthy Chinese subjects under fasting and fed conditions. Clin Pharmacol Drug Dev. 2020;9(3):346-352.
[Crossref] [Google Scholar] [PubMed]
- Pineyron E, Gomez M, Arellano M, Lopez E, Burke V, Gonzalez M. Bioequivalence Studies of Rivaroxaban at Two Different Strengths: Rivaroxaban 10 mg under Fasting Conditions and Rivaroxaban 20 mg under Fed Conditions in Healthy Mexican Subjects. Bioequiv & Bioavailab Int J. 2023;7(2):1-6.
[Crossref]
- Genisâ?Najera L, Sañudoâ?Maury ME, Moquete T. A singleâ?blind, randomized, singleâ?dose, twoâ?sequence, twoâ?period, crossover study to assess the bioequivalence between two oral tablet formulations of Rivaroxaban 20 mg in healthy Mexican volunteers. Clin Pharmacol Drug Dev. 2022;11(7):826-831.
[Crossref] [Google Scholar] [PubMed]
- Zografos L, Wolin D, Andrews E, Calingaert B, Balabanova Y, Horvat-Bröcker A, et al. Evaluating patient and physician knowledge of risks and safe use of Rivaroxaban: A survey across four countries. Expert Opin Drug Saf. 2022;21(3):435-446.
[Crossref] [Google Scholar] [PubMed]
- Ruigómez A, Schink T, Voss A, Herings RM, Smits E, Swart-Polinder K, et al. Safety profile of Rivaroxaban in first-time users treated for venous thromboembolism in four European countries. Plos one. 2024;19(3):0298596.
[Crossref] [Google Scholar] [PubMed]
Citation: Tjandrawinata RR, Yunaidi DA, Kurniawan YI, Nindyatami C, Saputro ID, Rahma IA, et al. (2024). Pharmacokinetic Equivalence Study of the Oral Direct Inhibitor of Factor Xa Anticoagulant, Rivaroxaban Film-Coated Tablets, in Healthy Subjects Under Fasting and Fed Conditions. J Bioequiv Availab. 16:595.
Copyright: © 2024 Tjandrawinata RR, et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.