
Indexed In
- Open J Gate
- Genamics JournalSeek
- Academic Keys
- JournalTOCs
- ResearchBible
- Ulrich's Periodicals Directory
- Access to Global Online Research in Agriculture (AGORA)
- Electronic Journals Library
- RefSeek
- Hamdard University
- EBSCO A-Z
- OCLC- WorldCat
- SWB online catalog
- Virtual Library of Biology (vifabio)
- Publons
- MIAR
- Geneva Foundation for Medical Education and Research
- Euro Pub
- Google Scholar
Useful Links
Share This Page
Journal Flyer

Open Access Journals
- Agri and Aquaculture
- Biochemistry
- Bioinformatics & Systems Biology
- Business & Management
- Chemistry
- Clinical Sciences
- Engineering
- Food & Nutrition
- General Science
- Genetics & Molecular Biology
- Immunology & Microbiology
- Medical Sciences
- Neuroscience & Psychology
- Nursing & Health Care
- Pharmaceutical Sciences
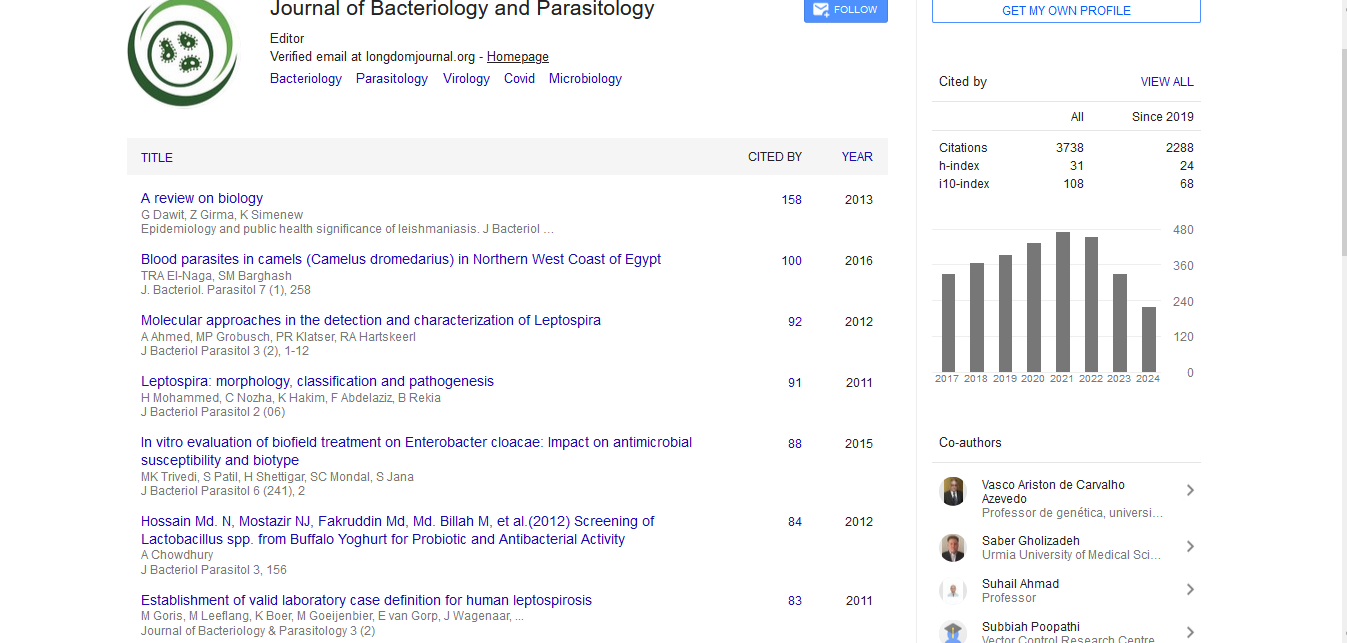
Research Article - (2020) Volume 11, Issue 4
Mortality Prediction for COVID-19 Patients: Methods and Potential
Peter Gemmar*Received: 07-Jul-2020 Published: 28-Jul-2020, DOI: 10.35248/2155-9597.20.11.374
Abstract
The pandemic spread of Coronavirus leads to increased burden on healthcare services worldwide. Experience shows that required medical treatment can reach limits at local clinics and fast and secure clinical assessment of the disease severity becomes vital. Biomarkers are regularly determined for intensive care patients. Machine learning tools can be used to select appropriate biomarkers in order to estimate the state of health and to predict patient mortality risk. Transparent prediction models allow further statements on the properties and development of the biomarkers in connection with specific health conditions of the intensive care patients.
In this work, alternative and advanced model approaches (Support Vector Machine, naive Bayes, Fuzzy system) are compared with models proposed in literature. In addition, aspects such as gender of patients and changes in biomarkers over time are included in the modeling. An artificial neural network (SOM) is used for selecting the biomarkers. A statistical analysis of the biomarkers reveals their values and changes in the critical state of the patients. In a model comparison, a Sugeno-type Fuzzy predictor achieved the best results for health assessment and decision support. The Fuzzy system delivers continuous output values instead of binary decisions and thus doubtful cases can be assigned to a rejection class. An extended Fuzzy model takes into account the patient’s gender and the trend in key features over time and thus provides excellent results with an accuracy better than 98% with the training data. However, this could not be finally verified due to the lack of suitable test data. The generation and training of all models was fully automatic with Matlab© tools and without additional adjustment.
Keywords
COVID-19 risk prediction; Computational intelligence; Supervised learning; Fuzzy inference system
Introduction
In the outbreak of COVID-19 pandemic causing severe health concerns and consequences for health care services worldwide has been described in a catchy way [1]. It is stressed that the severity of cases is putting medical services under great pressure. Furthermore, the importance of distinguishing patients that require immediate medical attention is described and that there is a lack of capacity to identify cases at imminent risk of death. Blood tests and the identification of relevant biomarkers are considered the basis for important applications in connection with COVID-19 patients, such as a disease prognosis or an assessment of the condition of a patient in the clinic [2]. So far, no prognostic biomarkers have been determined to estimate the patients risks.
Consequently, research group [1] analysed blood samples of 485 patients from the region of Wuhan, China [1]. Then, a state of the art machine learning algorithm was used to identify the most discriminative biomarkers. Most crucial biomarkers have been revealed through optimization of a supervised XGBoost classifier [3]. Three key features have been derived: lactic dehydrogenase (LdH), lymphocytes (Lymp%), and highsensitivity c-reactive protein (hs-CrP). A clinically operable decision tree (recTree) was developed and the decision rules with the three features as predictor variables and their thresholds were devised recursively by supervised learning.
The advantages of the recTree model are its simplicity and that it is easy to interpret, but it only delivers binary decisions and only offers orthogonal hyperplanes for the delimitation of predictor variables. There are many other possibilities for building a classification or prediction model [4]. We examined another three of them with very little effort for creation: 1st a Support Vector Machine (SVM), 2nd a naive Bayes classifier (nBayes), and 3rd a Sugeno-type Fuzzy classifier (Fis) [5]. All classifiers are transparent in explaining a specific input transformation to a specific classification output.
The classifiers SVM and nBayes delivered binary predictions at least as accurate as the classifier recTree. The classifier Fis is different from the three others as its output esteems the grade about how much the input belongs to one of two classes (positive, negative) specified as patient outcome in the data samples. This may be an advantageous property when predicting the patients risk value in practice.
There are also many possibilities for feature analysis and selection [6]. In our approach we put emphasis on finding those features that show signatures similar to the patient outcome and that are little to not correlated. Artificial neural networks of type Kohonen can be used to map the distribution of features in the feature space into 2D component planes (maps) revealing the signature of the according feature (Self Organizing Maps SOM) [7]. These maps can be compared visually and those maps similar to the map of patient outcome can be identified for feature selection. In addition, correlation analysis about the features can be used to determine the minimum feature set covering the feature space in an efficient way, e.g. in terms of a minimum dominant set (MDS). The key features selected in have been confirmed this way. Furthermore, two other features (Albumin, International Standard Ratio (ISR)) have been proposed and then used with the Fis classifier in an extended analysis.
If one looks at the determined biomarker values in the data base one will of course notice a change in the biomarkers from the days before the last sample was taken before discharge from the hospital [1]. In addition, an analysis of the statistical characteristics of the biomarkers shows that there are differences in the values for the two genders that advise separate use. It is therefore obvious to consider gender and the trend of the biomarkers in the risk assessment. This is successfully examined here with an expansion of the Fuzzy model. In this way, the Fis system enables a prediction of the mortality risk with an accuracy better than 98%. The time horizon can be up to approx. 20 days until the day of discharge, although this is not a forecast but a risk prediction based on the patient ’ s health condition before discharge. Ultimately, the Fis system shows potential for outstanding prediction results, although it must be said restrictively, that this has not yet been finally verified due to insufficient test data.
Data Resources
Basically, two data sets have been available for this study provided by [1]: trainData for training and external testData for testing or verifying the models [1]. trainData collects 74 biomarkers (features) together with age, gender, data sample time, admission time, discharge time, and class of patient outcome (alive, deceased) for 375 patients. testData collects three biomarkers LdH, Lymp%, and hs-CrP together with data sample time, admission time, discharge time, and class of patient outcome for 110 patients. Of the 375 patients in trainData, these three biomarkers are completely recorded in 351 patients. We have no biomarkers recorded continuously and uniformly, on the contrary, the biomarkers are incomplete and recorded at different times. To make matters worse, the data is not available as a time series with a time profile and fixed time intervals. The temporal horizon of any predictor below can only be estimated indirectly on the basis of the time delay between the date of the input data and a specified date (discharge date).
Only data of the final feature samples per patient is used for training and testing of the rule decision classifier recTree [1]. The distribution of patient outcome (positive: outcome deceased, and negative: outcome alive) over the space of the three features are depicted (Figure 1).

Figure 1: Patient outcome in train Data (left) and in test Data (right).
Feature and Data Analysis
Feature analysis can be carried out with different objectives: a) selection of the features for best prediction results, or b) selection of features for an optimal description of the patient’s state of health. The results do not have to be the same but not contradictory. Feature analysis resulted in determination of three features LdH, Lymp%, and hs-CrP out of 10 most promising features found with optimal XGBoost classifier output [1]. Our approach looks for features that correspond in their distribution to the patient’s outcome and then selects a minimum number of features that cover the input space well. The biomedical relevance of the biomarkers was initially not taken into account, but the selection found makes it possible to draw conclusions about the biomedical relevance and the relationship of the biomarkers to the prediction goal.
Here, feature analysis is accomplished in two steps: 1) a Kohonen neural network (SOM) is used for transforming the feature data into component planes CP, and 2) a Greedy algorithm is used for finding the minimum dominant set MDS of features based on their mutual correlation. Both, CP and MDS can be rendered and visually inspected. Figure 2 shows the component maps of 10 features selected in [1] after training SOM with 344 complete data samples in train data. The map size of SOM was 8x3=24 neurons (Figure 2).

Figure 2: Component planes CP (map size [8,3]) of 10 features (biomarkers) and patient outcome created by training SOM network with train data.
The component planes represent the weights of the respective feature in each neuron (hexagon) of the SOM map [8]. Each map position (hexagon) represents the weight value in color, and together with its neighbours around it corresponds with similar inputs (feature vectors) of the training set. A good approach for analyzing is to look for boundaries and color changes in a component plane and similar situations in other planes (colors must not be similar). In this way we recognize good matches in Figure 2 between outcome, Lactate dehydrogenase, (%) lymphozyte, Hypersensitive c-reactive protein, and with some restrictions also albumin and International standard ratio. The first three features correspond to the features selected for recTree and replicate the results of XGBoost classifier.
The second step of our feature analysis is to find the minimum dominant set MDS of features. For this, the mutual correlation of feature elements are evaluated and a greedy algorithm searches MDS after determination a threshold for mutual feature correlation [10]. Figure 3 shows the resulting correlation graph. We see the features Lactate dehydrogenase, (%) lymphozyte, and Hypersensitive c-reactive protein cover very well the MDS when we restrict it to the features with good matches with patient outcome. A strictly reduced MDS would consist only of (%) lymphozyte, and Hypersensitive c-reactive protein (Figure 3).

Figure 3: Correlation graph (lines show mutual correlation with threshold=0.95).
Feature distribution aspects
When selecting a suitable classifier method, both the distribution of the elements in the feature space and the classconditioned distribution of the feature values themselves are important. We now consider the latter based on the patient’s outcome class. Figure 4 shows the histograms of the three features for the last samples in train Data. And accordingly Figure 5 shows this for testData. The features are neither uniformly distributed nor disjoint, which may influence the choice of the predictor method and parameters.

Figure 4: Histogramm of main biomarkers LdH, Lymp%, and hs-CrP (from left to right) in trainData (last sample).

Figure 5: Histogramm of main biomarkers LdH, Lymp%, and hs-CrP (from left to right) in testData (last sample).
Gender aspects
Biomarker values are usually different for women and men. Accordingly, we also consider the values of the three biomarkers separately by gender in testData; trainData does not contain patient gender information. There are similarities in the distribution but also certain differences in the numerical values of the biomarkers with regard to the two outcome classes (please refer to the supplements for details). This becomes clear quantitatively when one compares the statistical values mean and standard deviation of the biomarkers (Table 1). These differences can also be seen in the values of the biomarkers immediately after admission to the hospital (Table 2). We can see a trend in biomarkers from the day of admission to the day of discharge: LdH increases on average for the deceased and decreases for the survivors, Lymp% decreases for the deceased and hs-CrP decreases for the survivors.
| Biomarker | LdH | Lymp% | hs-CrP |
|---|---|---|---|
| mean ± std | mean ± std | mean ± std | |
| Female alive | 208.6 ± 60.1 | 26.8 ± 10.0 | 26.8 ± 10.0 |
| Female deceased | 727.2 ± 320.0 | 5.5 ± 2.9 | 113.0 ± 72.7 |
| Male alive | 218.9 ± 70.2 | 25.1 ± 10.3 | 10.3 ± 19.9 |
| Male deceased | 803.0 ± 469.2 | 6.1 ± 6.8 | 6.1 ± 6.8 |
Table 1: Statistical values mean and standard deviation (STD) of selected biomarkers for women and men in trainData (last sample).
| Biomarker | LdH | Lymp% | hs-CrP |
|---|---|---|---|
| mean ± std | mean ± std | mean ± std | |
| Female alive | 246.6 ± 83.7 | 26.7 ± 10.7 | 25.8 ± 36.4 |
| Female deceased | 550.4 ± 256.1 | 7.6 ± 4.9 | 118.0 ± 106.6 |
| Male alive | 276.7 ± 134.0 | 22.1 ± 11.5 | 44.9 ± 47.9 |
| Male deceased | 622.1 ± 394.8 | 14.5 ± 77.8 | 147.6 ± 190.5 |
Table 2: Statistical values mean and standard deviation (std) of selected biomarkers for women and men in trainData immediately after admission.
Building a Prediction Model
Various methods are available for developing a predictor model [4]. In principle, one can differentiate between deterministic and probabilistic models. The former calculate nominal values as a membership degree of an input element X belonging to a class c, the latter calculate the posterior probability with which an input element X belongs to a class c, which makes a big difference. It is also important to consider whether the objective is to identify a trend or a fact. Furthermore, a distinction can be made between models with categorical and continuous output values.
A recursive decision tree (recTree) is chosen as the predictor model in [1]. With three input variables and associated threshold values, the input space is divided by means of orthogonal discriminating hyperplanes into two class regions. The output is categorical (binary) and therefore an input element X is always assigned with the membership to one of the two possible classes c positive, or negative . A trend in the input data is not considered with this model. The advantage of recTree is its simplicity and interpretability, which can be advantageous for practical use in a decisionmaking process. A disadvantage is the lack of plasticity in the linear decision hyperplanes in the case of an inhomogeneous distribution of the training samples.
Other nonlinear geometric and categorical predictor models such as Support Vector Machine SVM or k-nearest neighbour kNN are much more flexible, but unfortunately not as transparent and therefore more difficult to interpret. With knearest neighbour, assignments to similar patterns from the training set and thus classifications can be made. Initial tests with kNN were not promising and are therefore not considered further. With SVM, the margin between the support vectors of both classes that are equally closest to a decision boundary D(X) can be maximized. Input vectors X are thus assigned to the class depending on their position above or below the decision boundary D (D(X)>0 or D(X)<0). In cases of overlapping classes soft margin hyperplanes can be used that separate many but not all data points X.
The class-conditioned distribution of the three biomarkers is not disjoint, as can easily be seen in Figure 1. This raises the question of whether a probabilistic predictor model is better suited. This would then provide an indication of how likely an element belongs to either class positive or negative, but not to what degree the element belongs to a class due to its biomarkers. A possible probabilistic model can be created quite simply with a so-called naive Bayes model. For this purpose, the multivariate distribution of the input data points X is modeled and an input element X is assigned to class c, which has the greatest posterior probability, depending on the prior class probability (Bayes’ Rule). But who do you trust more in practice, the likelihood or the degree of class membership?
So the objective remains still to find a transparent model for the medical decision making process that is sufficiently adaptable, does not necessarily provide a binary decision and can also take temporal developments into account. A Fuzzy model Fis is recommended. We assume a human decision maker would prefer to refer to the technical estimation of risk grades when finally deciding about the risk and clinical treatment of patients. Furthermore, the mapping of biomarkers by humans likely will rather be in terms like small, high, or something unsharp like that than in sharply defined intervals. Fuzzy systems enable the description of models based on Fuzzy rules of type Ri: IF x1 is small AND x2 is large, THEN yi is small with input vector X in the premise (IF) part and output yi in the conclusion (THEN) part [9]. Building up a Fuzzy model requires first the definition of unsharp terms like small, medium, so called Fuzzy terms, covering the input elements and second the generation of the rule base describing the complete mapping of the input space into the output function. Finally, the mapping of the rule ’ s outputs yi into a sharp output value y, has to be established. Fortunately, there are a lot of machine learning tools that can automatically generate an operable Fuzzy model from training data.
We used a Sugeno-type Fuzzy model with linear functions yi=f(X) in the conclusion part and Matlab© function ANFIS for generating and training of the Fuzzy model. With trainData and three features as input ANFIS creates a Fuzzy model Fis with three Fuzzy terms per feature and N=33=27 rules. The model is trained by ANFIS with 10 epochs and trainData with all 351 patient’s final data samples only. Figure 6 shows prediction results of Fis for validation with trainData and verification with external testData.

Figure 6: Prediction results of Fis classifier validated with train Data (left) and tested with test Data (right); false classified data points circled.
Estimation Of Prediction Output
Some details on the development of the Fis model are described below. First, however, there is a performance comparison of the four model approaches recTree, SVM, nBayes, and Fis with the training data trainData and test data testData. The performance comparison consists of two steps (except recTree defined in [1]): 1) modeling with 10-fold cross-validation from trainData (always patient’s last sample), and 2) testing the models with the fewest total errors in 10-fold cross-validation with trainData or testData completely. For evaluation of the classifier results the following performances measures have been considered: total number of classification errors E, sensitivity = TP/(TP+ FN), specificity = TN/(TN+FP), and accuracy = (TP+TN)/(TP+TN +FP+FN); TP, TN, FP and FN stand for true positive, true negative, false positive and false negative rates.
The four models described above delivered the same results when tested with three biomarkers from testData, which is primarily attributable to the distribution of the test events (Table 3). In cross-validation, however, the Fuzzy model Fis delivers better results than the other models.
| recTree | SVM | nBayes | Fis | |||||
|---|---|---|---|---|---|---|---|---|
| Validation | Test | Validation | Test | Validation | Test | Validation | Test | |
| Errors total | 9 | 3 | 8 | 3 | 10 | 3 | 5 | 3 |
| Sensitivity (%) | 98.1 | 92.3 | 98.1 | 92.3 | 98.1 | 92.3 | 98.7 | 92.3 |
| Specificity (%) | 96.9 | 97.9 | 96.9 | 97.9 | 96.4 | 97.9 | 98.4 | 97.9 |
| Accuracy (%) | 97.4 | 97.3 | 97.4 | 97.3 | 97. 2 | 97.3 | 98.6 | 97.3 |
Table 3: Performance data of models during validation with trainData (351 samples) and verification (test) with testData (110 samples).
We know the appropriate data representation often improves the effectiveness of a model more than different methods for model building. For this reason, we also considered interacting data in addition to the selected biomarkers. We examined the effect of the interacting features gender and age on the main biomarkers Lymp%, LdH, and hs-CrP. Since age and gender are not included in testData, we performed a 10-fold cross-validation with trainData and selected the best model than for testing with complete trainData. These investigations were only possible with the SVM, nBayes, and Fis models. For the comparison, the models are trained with standard settings for the task and without manual optimization in order to recognize the basic potential. Even if the accuracy of the models was not significantly improved with the addition of a further biomarker and or the patients ’ age, the Fuzzy predictor here reaches a specificity of 100%. However, this was also achieved by considering the patients’ gender with the Fuzzy predictor and three biomarkers. The influence is also dependent on the predictor model, whereby it should be noted that with the addition of the interacting features, the models become more complex and the size of the training data can become too small for the supervised learning (see supplementary information for details).
Even as a medical layperson, it can be assumed that the patient’s physiological state and the health risk can also be judged by the development of the biomarkers over time and not only by their last value. Therefore, we tried to include the biomarkers’ trend in time into the model for risk assessment. However, it is now the case here that the blood samples and thus the biomarkers in the data records were not systematically recorded over time. Thus, it is only possible to determine the temporal trend here as an example. To do this, we simply chose the gradient as weighted difference between the last and penultimate data sample as a measure for the trend over time. The results were positive for all models and with ideal results for Fis (see supplementary information for details). The very good results of all models with the gradient of a biomarker are to be judged cautiously, because ideal overfitting cannot be ruled out because of the relatively small number of training samples. Nevertheless, it remains a clear indication that the gradient of the biomarkers is an important feature for mortality risk prediction.
In comparison of the models, the Fuzzy Model Fis has a high degree of flexibility and provides the best performance data. In addition, the primary model output specifies the degree to which an input can be assigned to class positive, for example, and thus offers the possibility for further interpretations. Since there is a two-class problem here, a rejection class critical can be introduced for model issues in the disputed area, for example for degrees around 0.5±0.1. Figure 7 shows the results of the Fis classifier with two classes plus rejection and how rejected elements are distributed in the input space.

Figure 7: Distribution of patient samples in trainData (left) and testData (right) classified with Fis in two classes positive, negative plus rejection critical; false classifications with circles.
This supports an interpretation of the model output by medical professionals. If one looks at the real risk values that model Fis calculates for the external test data, one can see that the wrongly classified items in most cases are very close to the decision limit θ=0.5. Figure 8 shows the statistical values of the risk assessment with the training and test data using boxplots. After assigning the incorrectly classified examples (FN=2, FP=1 in Figure 7), the associated boxplot shows that the real output values are close to the decision value θ and can therefore be better assessed by a medical expert than the wrong binary decision. The continuous model output enables further opportunities for technical support for medical experts in COVID-19 risk assessment.

Figure 8: Statistical distributuion of risk assessment (real model outputs) with prediction model Fis and training data trainData (left) and test data testData (right), boxplots with 25, 75 percentil (blue), median and outliers (red).
Time horizon
With regard to the prediction horizon, it was already criticized above that there is no actual time series for the biomarkers and that a trend cannot be determined uniformly. In order to be able to make statements about the forecast horizon under this restriction, the time delays between the classified facts and the discharge date can be considered. For this purpose, the Fis model with three input features LdH, Lymp%, and hs-CrP was used to predict the mortality risk of the last and penultimate samples from trainData and determine the time delay from the recording date of the biomarkers (all three markers recorded at the same day) to the discharge date. Figure 9 shows the distribution of the correct and incorrect risk prediction values with regard to the time delay and also the relative accuracy of the prediction. However, this consideration can only be interpreted as how many days before the discharge the patients had reached a state of health so that it could be decided how the discharge would be (deceased or alive). This should not be confused with a forecast, because it would say something about the future development of the patient ’ s health status and ultimately about the expected discharge status.

Figure 9: Histogram of the correct and incorrect mortality risk values estimated in days before the discharge and the corresponding accuracy with Fis with three inputs LdH, Lymp%, and hs-CrP; days are the time interval between the recording date of the data and the patient’s discharge date.
Fis model characteristics
The Fis model is represented by its rule base, making it transparent and relatively easy to interpret. Figure 10 shows a rule base with N=33=9 rules for a Fis model with two input variables LdH and Lymp% (reduction of model complexity for ease of presentation). The resulting decision surface of the predictor model is also shown (Figure 10).

Figure 10: Rule base (left) and decision surface (right) of Fis model with two input variables LdH and Lymp% and N=9 rules.
It is a practical experience to build models with as few rules as possible and to achieve the desired plasticity. In purely automated approaches, which is the case here, all the input variables are used with a uniform number of membership functions in the rules. If we use three biomarkers as input variables and define three membership functions for each variable, we get a rule base with rules. Each membership function is described by a parameter set, e.g. three parameters for generalized bell-shaped membership function (Matlab© glbellmf). The output of a rule is formed by a linear combination of the input variables and then summarized linearly over all rules (e.g. weighted average). This results in J=N(3+1)+N=135 parameters in total in this example. These parameters are optimized in an adaptive neural learning process (Matlab© ANFIS). From personal experience, it also applies here that at best about 10 training samples should be available for each parameter. This would require, for example, 1,350 training examples in the training set trainData. However, since there are significantly fewer training samples (351) and still very good results achieved, it cannot be ruled out that the models are overfitted to the training examples. This problem is counteracted during training with 10-fold cross-validation. The limits can also be seen from the fact that Fis models with more input variables do not achieve better results, because already a Fis model with four input variables generates a rule base with N=81 rules and J=441 parameters in total. trainData is clearly too small for automatic model optimization with more than three input variables.
Discussion
This study shows the potential of Fuzzy models for the mortality risk assessment of COVID-19 intensive care patients in several ways. First of all, the results of Fis are continuously better than in literature and no other of the methods considered here achieved better results. The Fis system also enables non-binary risk assessments and thus a differentiated assessment of the patient ’ s health condition. The consideration of patients ’ gender as an interacting feature improves the prediction performance, whereby it should be noted when developing the model that only about half of the size of the training data is still available.
The consideration of the temporal development of the biomarkers in the models had a decisive influence on the model performance. However, this could not be tested in detail because the training and external test data contained too few examples and in particular the blood samples had not been recorded systematically over time. For further model examination based on these positive results, more and systematically recorded training and test data are absolutely necessary.
The Fis model is transparent and its decision is easy to convey in an operational model application. No special optimization options were used here. However, it can be assumed that with specially selected and dimensioned Fuzzy membership functions for the input data, a simplification of the system can be achieved with the same performance.
Conclusion
In summary, this study compares a Fuzzy logic based prediction system for COVID-19 mortality risk assessment of intensive care patients with other deterministic and probabilistic prediction methods evaluated here or in literature. It could be shown, that Fuzzy logic based prediction delivers the best performance data in terms of accuracy, sensitivity and specificity. This provides a good basis for the development of a transparent and operational system for risk assessment of COVID-19 patients. It is advised, that patients’ gender and feature changes over time be integrated in the model input. The model output is non-binary and is therefore particularly suitable for a decisive interpretation by medical experts. An investigation of the time horizon was carried out to the extent that the time from the critical patient condition to discharge was determined. There is still great potential here for further investigations into the course of the medical features up to the discharge of the patients, for which patient data are required as suitable time series. This also includes the development of a prediction model for COVID-19 patients with mild symptoms.
Acknowledgements
I would like to thank the authors in for providing the training and test data [1].
Conflict of Interest
The author declares that he has no conflict of interest.
REFERENCES
- Yan L, Zhang H, Goncalves J, Xiao Y, Wang M, Guo Y, et al. An interpretable mortality prediction model for COVID-19 patients. Nature Machine Intelligence. 2020;2(1):283–288.
- Zadeh LA. Fuzzy sets. Information and Control. 1965;8(3):338-353.
- Tagaki T, Sugeno M. Fuzzy identification of systems and its applications to modelling and control. IEEE Trans Systems Man Cybernetics. 1985;15(1):116-132.
- SOM Toolbox 2.0. Laboratory of Computer and Information Science Adaptive Informatics Research Centre. 2015.
- Messner CB, Demichev V, Wendisch D, Michalick L, White M, Frei-wald A, et al. Ultra-High-Throughput Clinical Proteomics Reveals Classifiers of COVID-19 Infection. Cell Systems. 2020;11(1):1-14.
- Kuhn M, Johnson K. Feature Engineering and Selection: A Practical Approach for Predictive Models, CRC Press, Taylor & Francis Group, Boca Raton. 2020.
- Kohonen T. Self-Organizing Maps. (3rd edn) Springer, New York, USA. 2001. pp: 105-176.
- Kuhn M, Johnson K. Applied Predictive Modeling. (2nd edn) Springer, New York, USA. 2013. pp: 329-411.
- Chen T, Guestrin C. XGBoost: A Scalable Tree Boosting System. ACM. 2016;1(1):785-794.
- Faust C, Gemmar P, Gronz O, Casper M. Generating fuzzy logic-based rainfall-runoff models using self-organizing maps. The 20th Annual TIES Conference. 2009.
Citation: Gemmar P (2020) Mortality Prediction for COVID-19 Patients: Methods and Potential. J Bacteriol Parasitol. 11:374. DOI: 10.35248/2155-9597.20.11.374.
Copyright: © 2020 Gemmar P. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.