
Indexed In
- Open J Gate
- Genamics JournalSeek
- Academic Keys
- JournalTOCs
- The Global Impact Factor (GIF)
- China National Knowledge Infrastructure (CNKI)
- Ulrich's Periodicals Directory
- RefSeek
- Hamdard University
- EBSCO A-Z
- OCLC- WorldCat
- Publons
- Geneva Foundation for Medical Education and Research
- Euro Pub
- Google Scholar
Useful Links
Share This Page
Journal Flyer

Open Access Journals
- Agri and Aquaculture
- Biochemistry
- Bioinformatics & Systems Biology
- Business & Management
- Chemistry
- Clinical Sciences
- Engineering
- Food & Nutrition
- General Science
- Genetics & Molecular Biology
- Immunology & Microbiology
- Medical Sciences
- Neuroscience & Psychology
- Nursing & Health Care
- Pharmaceutical Sciences
Research Article - (2019) Volume 10, Issue 4
Molecular Docking Studies for Design, Synthesis and Characterization of New Imatinib Analogues
Ali Naim Hussein1, Omar F Abdul- Rasheed2*, Monther F Mahdi3 and Ayad MR Raauf42Department of Chemistry and Biochemistry, College of Medicine, Al-Nahrain University, Iraq
3Department of Pharmaceutical Chemistry, Ashur University College, Iraq
4Department of Pharmaceutical Chemistry, College of Pharmacy, Mustansiriyah University, Iraq
Received: 16-Oct-2019 Published: 22-Nov-2019
Abstract
Background: In silico drug design conducts a process for making conformations and directions of multiple ligands and chooses the best ones, then being selected. In silico studies are used to examine and model molecular interactions between target macromolecules and ligand. Tyrosine kinase is considered a potential target to design inhibitors. Tyrosine kinase inhibitor like imatinib this drug has succeeded to pass through clinical studies in an attempt to cure the cancer, which is considered as the second leading cause of deaths in the world. In this work, the GOLD program was employed to predict the bindings and thus the inhibitory activity toward the tyrosine kinase.
Methodology: After the design and docking processes, the chemical synthesis of three imatinib analogues was achieved.
Results: the percent of yield of the chemical synthesis was (81-85%). The synthesized compounds were well characterized using FTIR, NMR, DSC, and CHN elemental analyser. The present purity was within the globally accepted value of less than 0.4% with the employment of CHN combustion.
Keywords
Computer-aided drug design; In silico; Gold; Imatinib analogues; Anticancer; Tyrosine kinase inhibitor
Introduction
In 1989, researchers declared the term “in silico” to define biological test completely achieved on the computer or through simulation of computer [1]. Moreover, in silico drug design considers a great aspect in that various sides of practices and essential researches are combined and fill with each other [2]. This can be utilized to counteract cancer. Cancer is considered one of the most important health problems in the world, and that the rates of cancer incidence are high and come at the second level after heart diseases as the most important cause of death in America and all countries of the world. Currently, accounts of cancer 25% of deaths in the United States America [3,4].
Researchers noticed that abnormal activation of numerous receptor tyrosine kinases (RTKs) and respected ligands can cause uncontrolled proliferation of tumor cells which is pertinent to the events of the development of cancers IN human. Receptor tyrosine kinases establish a receptor family which monitoring the main cellular events, such as apoptosis, cell proliferation, migration and differentiation. The Receptor tyrosine kinases family comprises growth factor receptors, like the receptor of stem cell factor (SCF), the receptor of insulin growth factor, the receptor of epidermal growth factor, and PDGFR (the receptor of platelet-derived growth factor) [5].
In this study, the researchers aimed to design, synthesize, and characterize novel tyrosine kinase inhibitors with the utilization of the GOLD software version 5.7.2, obtained by CCDC (the Cambridge Crystallographic Data Centre).
Materials and Methods
Materials
Imatinib (99.8%) and N-(5-Amino-2-methylphenyl)-4-(3-pyridyl)-2- pyrimidineamine, (98%) were purchased from BLDpharm, China. Acetyl chloride, (98.0%) and Benzoyl chloride, A. R. (99.5%) were obtained from CDH, India. Dichloromethane, HPLC grade (99.8%) and n-Hexane, (95.0%) were purchased from GCC, UK. Tetrahydrofuran, A. R. (99.0%) and Potassium carbonate, A. R. (99.0%) were purchased from SCR, China. Methanol absolute, HPLC grade were gained from Biosolve ChimieSARL, France. Finally, Tetramethylacetyl chloride, (99.0%) were purchased from Sigma-Aldrich, USA.
Instruments
The used instruments in this study are listed as shown in Table 1.
| No. | Instruments | Manufacturers | Countries |
|---|---|---|---|
| 1 | Differential Scanning Calorimeter (DSC) Thermal Analyzer | Shimadzu | JAPAN |
| 2 | C.H.N Elemental Analyzer | EURO EA3000 | ITALY |
| 3 | Melting point apparatus | BioCote | UK |
Table 1: The employed instruments in the current research.
The in silico docking process
Preparation of proteins: The structure of X-ray crystallographicof the Abl kinase (PDB ID 3K5V) protein has been obtained from PDB (Protein Data Bank) at the resolution 1.74 Å (Figure 1). Then, the structure file was opened with ChemBio 3D Ultra 14 program, and the energy minimization step was performed. The energy-minimized protein structure was saved as a “mol2” file. The addition of atoms of hydrogen to the protein has been achieved using Mercury version 4.1.3. Thereafter, the mol2” file was reopened in the Hermes (GOLD) program. In the GOLD program, ligands, molecules of water, and other heteroatoms have been extracted from macromolecules of the protein.

Figure 1: The crystalline structure of Abl kinase in the complex plus GNF- 2 (PDB ID 3K5V), and imatinib.
Ligand preparation: From many others, three best fitted and chemical synthesis-capable imatinib analogues were designed and synthesized. These analogues are listed in Table 2. The structures of the ligands have been drawn in the software of ChemBio Draw Ultra ver. 14, while the energy minimization was achieved with the software of ChemBio 3D Ultra ver. 14. The energy-minimized protein and ligand have been saved in mol 2, and PDB formats, respectively.
| Code | IUPAC name | Formula | Chemical structure |
|---|---|---|---|
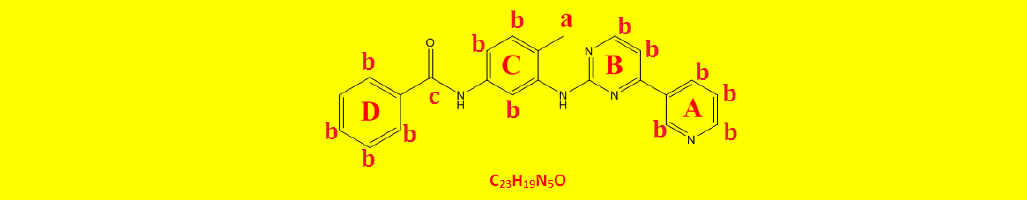
| 2a | N-(4-methyl-3-((4-(pyridine-3-yl)pyrimidin-2-yl)amino)phenyl)benzamide | C23H19N5O | 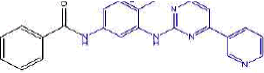 |
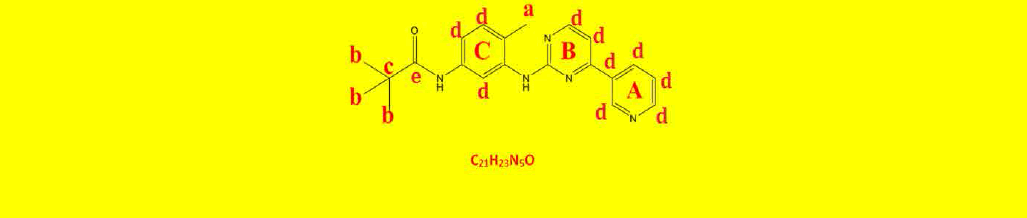
| 2b | N-(4-methyl-3-((4-(pyridine-3-yl)pyrimidin-2-yl)amino)phenyl)pivamide | C21H23N5O | 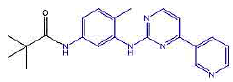 |
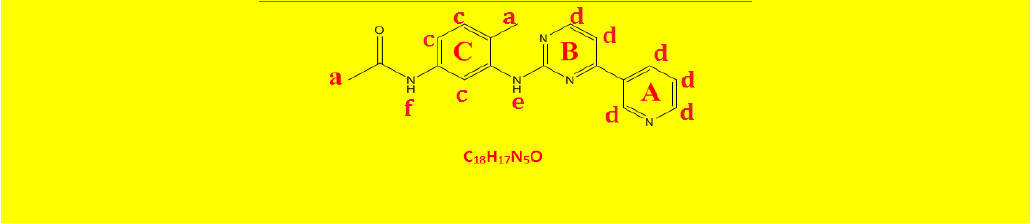
| 2c | N-(4-methyl-3-((4-(pyridine-3-yl)pyrimidin-2-yl)amino)phenyl)acetamide | C18H17N5O | 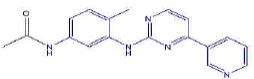 |
Table 2: The symbols, IUPAC names, chemical formulas, and the chemical structures of the synthesized imatinib analogues.
Molecular docking utilizing the GOLD software: The molecular docking was achieved using GOLD version 5.7.2. This was done to predict the scores of GOLD for interactions of the targeted protein–ligand. The sites of binding of the residues of amino acids of the defined proteins have been determined and applied for the software of GOLD. This was conducted for atoms with-in 10 Å of the submitted binding residues in the pocket of binding. However, the docked conformations that had the higher Score of GOLD (fitness) have been taken for analyzing the binding mode. Eventually, the program of GOLD has been run, and then solutions of GOLD have been observed, saved, and imagined.
The chemical synthesis: The overall chemical syntheses are revealed in Scheme 1 below [6]. Scheme 1 shows the chemical synthesis and conditions of the reaction of the compounds 2a, 2b, and 2c.
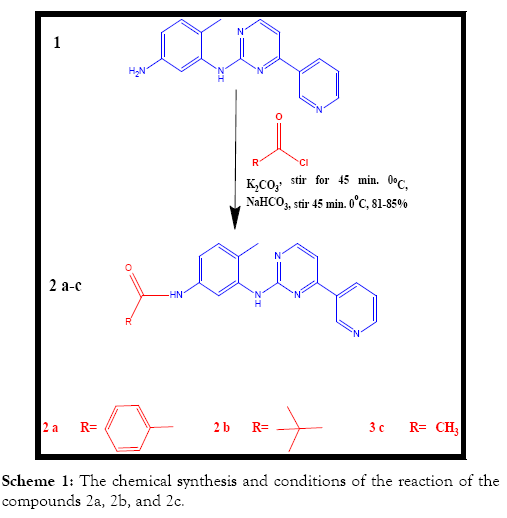
The following chemical approaches have been employed for the imatinib analogues preparations [7]:
Synthesis of compound (2a) N-(4-methyl-3-((4-(pyridine-3- yl)pyrimidin-2-yl)amino)phenyl)benzamide
About 309 mg of Benzoyl chloride (2.2 mmol) has been delivered to the stirred solution of 500 mg of the compound 1 (1.8 mmol) and 1.24 g of potassium carbonate (9 mmol) in 12 mL of THF at 0°C and the mixture of reaction has been stirred for 45 minutes at 0°C. The mixture of reaction has been delivered drop insight to the stirring, cold solution of 12 mL of 10% NaHCO3 and stirred for 30 min at 0°C. A solid precipitated out which has been isolated using filtration. The solid has been washed with cold water, hexane and dissolved in 10 mL of the mixture of dichloromethane/methanol (50:50,) and concentrated under the reduced pressure. The residue obtained has been suspended in 20 mL of cold water, Et3N has been added to it and it has been extracted twice with 20 mL of ethyl acetate. The combined ethyl acetate extract has been washed with 10 mL of water, concentrated under the reduced pressures within the desiccator for getting 2-a (0.568 mg, 82%).
Synthesis of compound (2b) N-(4-methyl-3-((4-(pyridine-3- yl)pyrimidin-2-yl)amino)phenyl)pivamide
About 266 mg of Trimethylacetyl chloride (2.2 mmol) has been delivered to the stirred solution of 500 mg the compound 1 (1.8 mmol) and 1.24 g of potassium carbonate (9 mmol) in 12 mL of THF at 0°C and the mixture of reaction has been stirred for 45 minutes at 0°C. The mixture of reaction has been delivered drop insight to the stirring, cold solution of 12 mL of 10% NaHCO3 and stirred for 30 min at 0°C. A solid precipitated out which has been isolated using filtration. The solid has been washed with cold water, hexane and dissolved in 10 mL of the mixture of dichloromethane/ methanol (50:50,) and concentrated under the reduced pressure. The residue obtained has been suspended in 20 mL of cold water, Et3N has been added to it and it has been extracted twice with 20 mL of ethyl acetate. The combined ethyl acetate extract has been washed with 10 mL of water, concentrated under the reduced pressures within the desiccator for getting 2-b (0.556 mg, 85%).
Synthesis of compound (2c) N-(4-methyl-3-((4-(pyridine-3- yl)pyrimidin-2-yl)amino)phenyl)acetamide
About 173 mg of Acetyl chloride (2.2 mmol) has been delivered to the stirred solution of 500 mg of the compound 1 (1.8 mmol) and 1.24 g of potassium carbonate (9 mmol) in 12 mL of THF at 0°C and the mixture of reaction has been stirred for 45 minutes at 0°C. The mixture of reaction has been delivered drop insight to the stirring, cold solution of 12 mL of 10% NaHCO3 and stirred for 30 min at 0°C. A solid precipitated out which has been isolated using filtration. The solid has been washed with cold water, hexane and dissolved in 10 mL of the mixture of dichloromethane/methanol (50:50) and concentrated under the reduced pressure. The residue obtained has been suspended in 20 mL of cold water, Et3N has been added to it and it has been extracted twice with 20 mL of ethyl acetate. The combined ethyl acetate extract has been washed with 10 mL of water, concentrated under the reduced pressures within the desiccator for getting 2c (0.463 mg, 81.0%).
Results and Discussion
The docking
Regarding the discipline of computer-aided drug design, the energy minimization (energy optimization, aka, geometry minimization, and geometry optimization) indicates the recoding of the spatial geometry of the atoms group in which the net energy of the origin of inter atomic on all atoms was near 0 [8-11]. However, the minimization of the energy was critical to observe trustable results of the docking and is achieved by finding the paradigm of receptor sites and ligands that must interact with it with the aid of a computer [12-18]. However, in the current study, energy minimizations have been registered for both the ligand and the macromolecule (i.e., the Abl kinase (PDB ID 3K5V) protein) to be used after that in the docking process.
Docking of the structures
The computational docking analyses have been achieved by GOLD software driven by CCDC (the Cambridge Crystallographic Data Centre) (www.ccdc.cam.ac.uk) besides other software.
The protein of interest is Abl kinase (PDB ID 3K5V). Imatinib plays as a tyrosine kinase inhibitor (TKI). This is true for the analogues; 2c, 2a, and 2b. Nevertheless, no constraints have been utilized in the docking processes. Regarding the site of binding, these have been determined as (GLU288, HIS362, and ASP382).
The analyses of the molecular docking were successfully achieved with the usage of the correlated drug design softwares. The rate of the successfulness for the GoldScore observed for the program of GOLD has been assigned as 57% [19]. However, in this work, the success rate was calculated to be 67%. Accordingly, the chemical compound 2a reflects the best GoldScore alongside the best fitting within the active site (better than imatinib) as revealed in Table 3.
| Chemical | Bound amino acids | GoldScore | H-bonds |
|---|---|---|---|
| Imatinib | 3 | 83.1 | 2 |
| 2a | 3 | 89.76 | 2 |
| 2b | 3 | 83.6 | 2 |
| 2c | 3 | 79.8 | 2 |
Table 3: Tested chemicals, the bound amino acid residues, the GoldScore, the number of the hydrogen bonds. Imatinib is utilized as a positive control.
Table 3 reveals the tested chemicals, the bound amino acid residues, the GoldScore, the number of the hydrogen bonds. Imatinib is utilized as a positive control. Docking of imatinib structure is shown in Figure 2. Docking of compound 2a is shown in Figure 3. Docking of compound 2b is exhibited in Figure 4. Docking of compound 2c is shown in Figure 5.

Figure 2: The crystal structure of imatinib in the complex with tyrosine kinase (PDB code: 3K5V). The tyrosine kinase is represented as the space fill model, while the imatinib is depicted as the capped stick model.

Figure 3: The crystal structure of compound 2a in the complex with tyrosine kinase (PDB code: 3K5V). The tyrosine kinase is represented as the space fill model, while the compound 2a is depicted as the capped stick model.

Figure 4: The crystal structure of compound 2b in the complex with tyrosine kinase (PDB code: 3K5V). The tyrosine kinase is represented as the space fill model, while the compound 2b is depicted as the capped stick model.

Figure 5: The crystal structure of compound 2c in the complex with tyrosine kinase (PDB code: 3K5V). The tyrosine kinase is represented as the space fill model, while the compound 2c is depicted as the capped stick model.
The chemical synthesis
Three analogues of imatinib have been manufactured and some of physical and chemical characteristics of the synthesized compounds are listed in Tables 4 and 5.
| Symbols of Compounds | The physical characteristics | DSC-Measured Melting Point (°C) | Calculated-tPSA* | Calculated-Log P |
|---|---|---|---|---|
| Imatinib (STI-571) | White powder | 213.42 | 75.13 A2 | 2.52 |
| 2a | Fluffy white-amber powder | 177.02 | 78.21 A2 | 4.13 |
| 2b | Crystalline white-amber powder | 185.13 | 78.21 A2 | 4.16 |
| 2c | Fluffy amber powder | 219.82 | 78.21 A2 | 2.23 |
tPSA*: The topological polar surface area is the summation of the surface of the totally polar atoms, firstly N and O and atoms of the attached H.
Table 4: The physical characteristic of imatinib standard and analogues of the recently synthesized compounds.
| The symbol of compounds | The chemical formulae | Molecular mass | Yield Percentage | Donor of H-Bond | Acceptor of H-Bond | Count of Rotatable bonds | Heavy atoms count |
|---|---|---|---|---|---|---|---|
| STI-571 | C29H31N7O | 493.60 | STD | 2 | 8 | 8 | 37 |
| 2a | C23H19N5O | 381.44 | 82 | 2 | 6 | 5 | 29 |
| 2b | C21H23N5O | 361.45 | 85 | 2 | 6 | 8 | 27 |
| 2c | C18H17N5O | 319.37 | 81 | 2 | 6 | 5 | 24 |
Table 5: The chemical parameters of imatinib standard and the analogues of recently synthesized compounds.
The observed data within Tables 5 and 6 are considered fruitful. First of all, the observed melting points were obtained with both DSC and melting point apparatus. It is a good point to mention that the DSC has priority in the detection of the compounds’ melting point. This is obvious as the sensitive Shimadzu’s machine gives the required data within two decimals. This, in particular, cannot be inquired with the classical capillary apparatus. The DSC has the ability to register the enthalpies and transition temperatures. This in turn enhances the fact that the DSC has a valuable capability to observe a very sensitive and characteristic melting point for the target chemical compound [20]. In this work the melting points were observed for imatinib and the other chemicals.
| Compound symbol | Chemical formula | Molecular mass | Elemental microanalyses % | ||
|---|---|---|---|---|---|
| Element | Observed | Calculated | |||
| STI-571 | C29H31N7O | 493.60 | C | 70.179 | 70.28 |
| H | 6.599 | 6.71 | |||
| N | 19.693 | 19.78 | |||
| Sum | 96.471 | 96.77 | |||
| % Deviation | 0.30% | ||||
| 2a | C23H19N5O | 381.44 | C | 72.290 | 72.42 |
| H | 4.920 | 5.02 | |||
| N | 18.254 | 18.36 | |||
| Sum | 95.464 | 95.8 | |||
| % Deviation | 0.35% | ||||
| 2b | C21H23N5O | 361.45 | C | 69.598 | 69.78 |
| H | 6.398 | 6.41 | |||
| N | 19.274 | 19.38 | |||
| Sum | 95.27 | 95.57 | |||
| % Deviation | 0.31% | ||||
| 2c | C18H17N5O | 319.37 | C | 67.598 | 67.70 |
| H | 5.282 | 5.37 | |||
| N | 21.872 | 21.93 | |||
| Sum | 94.752 | 95 | |||
| % Deviation | 0.26% | ||||
Table 6: imatinib and the synthesized compounds elemental microanalyses.
Furthermore, the observed log P (the partition coefficient of octanol-water) values, the acceptors of H-bond, and the donors of H-bond show a suitable submissiveness for the famous rule of 5. This rule of five (Lipinski rule of five) states that the drug candidate has the probability to be an orally vigorous just if it possesses no more than 1 breach of the below [21-25]:
i. H-bond acceptors must be less than ten (the sum of atoms of O and N).
ii. H-bond donors must be less than five (the sum of bonds of O–H and N–H).
iii. The molecular mass must be less than 500 grams per mole.
iv. The log P must be less than five units.
Moreover, to assure the oral absorption of a drug candidate, the count of the rotatable bonds has to be considered. No more than ten rotatable bonds have to be exist for the chemical compound to be orally available. In the same context, the tPSA has to be no more than 140 A2 [26,27]. The tPSA is commonly used parameter in medicinal chemistry to assure the cell permeability by the drug candidate [28-30].
On the other hand, to estimate the reaction efficiency, the percent of yield has to be calculated. Simply, the percent of yields is determined as ratio of the amount of the product that is gained in a chemical reaction to the theoretically calculated amount multiplied by 100 [31]. Notably, all the term very good is correlated with the percentage yields of the chemical synthesis of the current research [31,32].
The properties of synthesized compounds
Infrared spectroscopy (IR): In the drug analyses, the spectrum of IR is a rapid, reliable, non-destructive commonly used technique [33]. The amide carbonyl can be easily characterized with the employment of the FT-IR tool [34]. In this work, the characterization of the amide carbonyl was conducted for the observed spectra.
Melting points acquisition: Melting points were acquired with the aid of Differential Scanning Calorimeter (DSC) Thermal Analyser. In this work, the observed melting points were gained with a sensitivity of two decimal places which has the superiority to the traditional melting point tester apparatus.
The elemental micro-analysis (CHN): The elemental micro analysis is a cornerstone in the characterization of the chemical compounds. This technique relies on the contents of elements in the compound. This encompasses the carbon (C), hydrogen (H), and nitrogen (N) or so-called (CHN) which are the elements that the analyser of elements is capable to detect [35]. Fundamentally, deviation of the observed weights to those theoretically calculated must not exceed the 0.4%. this is a must to assure the purity of the chemical product [35,36].
In this work, the calculated deviations of the weights of the synthesized chemicals were less than the worldwide agreed value of 0.4%. This indicates low contents of impurities, as seen in Table 6.
Proton NMR
For the structure elucidation, it globally agreed that the proton NMR is the cornerstone in this field to show the hydrogen atoms map [37-41].
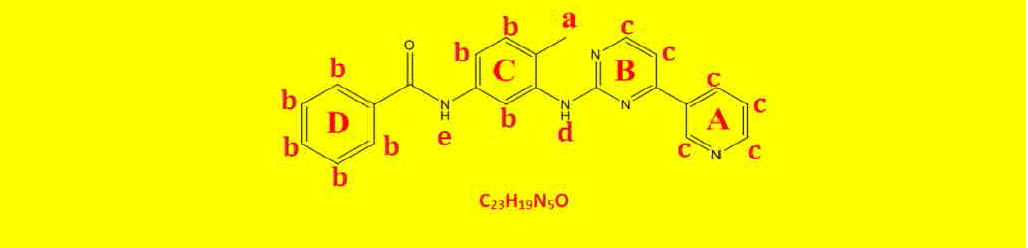
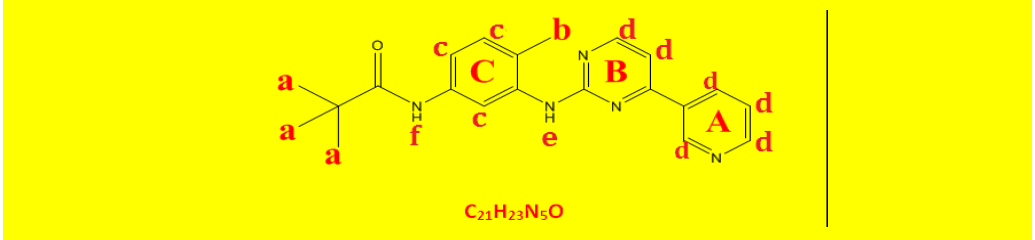
The proton NMR explanation of compound 2a is shown in Table 7. The proton NMR explanation of compound 2b is shown in Table 8. The proton NMR explanation of compound 2c is shown in Table 9.
 |
||||
|---|---|---|---|---|
| Chemical group | Chemical Shift (ppm) | Integrations | No. of Hydrogens | Interpretations |
| a | 2.27 | 5.69 | 3 | Singlet, for CH3 protons |
| b | 7.23-8.02 | 15.61 | 8 | Aromatics ring C and D. |
| C | 7.99-8.71 | 11.96 | 6 | Hetroaromatic ring A and B |
| d | 9.32 | 1.94 | 1 | Singlet, for N-H proton. |
| e | 10.28 | 1.85 | 1 | Singlet, for N-H proton. |
| Sum=37.05 | Total=19 | 37.05 ÷ 19=1.95 integrations per proton | ||
Table 7: Proton NMR interpretations of compound 2a.
 |
||||
|---|---|---|---|---|
| Chemical group | Chemical Shift (ppm) | Integrations | No. of Hydrogens | Interpretations |
| a | 1.31 | 8.95 | 9 | Singlet, for three CH3 groups of the tert-butyl protons. |
| b | 2.23 | 2.83 | 3 | Singlet, for CH3 protons |
| c | 7.15 | 7.53 | 3 | Aromatics ring C. |
| d | 8.01 | 8.95 | 6 | Hetroaromatics A and b. |
| e | 9.18 | 0.94 | 1 | Singlet, for N-H proton of amide. |
| f | 9.29 | 1.00 | 1 | Singlet, for N-H proton of amide. |
| Sum=22.99 | Total=23 | |||
Table 8: Proton NMR interpretations of compound 2b.
 |
||||
|---|---|---|---|---|
| Chemical group | Chemical Shift (ppm) | Integrations | No. of Hydrogens | Interpretations |
| a | 2.07 | 2.93 | 3 | Singlet, for CH3 protons |
| b | 2.22 | 2.90 | 3 | Singlet, for CH3 protons |
| c | 7.14 | 7.54 | 3 | Aromatics ring C . |
| d | 7.92 | 8.97 | 6 | Hetroaromatics A and b . |
| e | 9.18 | 1.02 | 1 | Singlet, for N-H proton. |
| F | 9.29 | 0.91 | 1 | Singlet, for N-H proton. |
| Sum=17 | Total=17 | |||
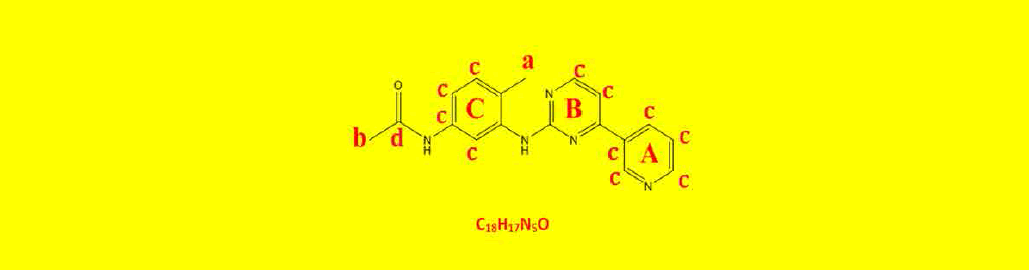
Table 9: Proton NMR interpretations of compound 2c.
In the current study, the NMR of proton has been successfully utilized to determine hydrogen atoms in the chemically synthesized compound.
Carbon-13 NMR
Certainly, the carbon-13 NMR is considered as an important technique used in the synthetic organic chemistry discipline to determinate the chemical structure of the target. In fact, carbon-13 NMR is correlated with the elucidation of carbon atoms in the chemical compound [42-44]. Consequently, carbon-13 NMR has been employed happily to elucidate the chemical structure of the synthesized chemical compound. The carbon-13 NMR interpretation of compound 2a is revealed in Table 10. The carbon-13 NMR interpretation of compound 2b is revealed in Table 11. The carbon-13 NMR interpretation of compound 2b is revealed illustrated in Table 12.
 |
|||
|---|---|---|---|
| Chemical group | Chemical Shift (ppm) | No. of Carbons | Interpretations |
| A | 18.16 | 1 | For the methyl CH3 carbon. |
| B | 108.00-162.10 | 21 | Aromatics, heterocyclics; aromatic. |
| C | 165.89 | 1 | For carbonyl carbon. |
| Total=23 | |||
Table 10: Carbon-13 NMR interpretations of compound 2a.
 |
|||
|---|---|---|---|
| Chemical group | Chemical Shift (ppm) | No. of Carbons | Interpretations |
| A | 18.05 | 1 | For the methyl CH3 carbon. |
| B | 27.74 | 3 | For the three methyl CH3 carbons. |
| C | 39.58 | 1 | For the central carbon of the tert-butyl moiety. |
| D | 107.91-161.99 | 15 | Aromatics, heterocyclics; aromatic. |
| E | 176.73 | 1 | For carbonyl carbon. |
| Total=21 | |||
Table 11: Carbon-13 NMR interpretations of compound 2b.
 |
|||
|---|---|---|---|
| Chemical group | Chemical Shift (ppm) | No. of Carbons | Interpretations |
| A | 18.08 | 1 | For the methyl CH3 carbon. |
| B | 24.48 | 1 | For the methyl CH3 carbon. |
| C | 107.98-162.08 | 15 | Aromatics, heterocyclics; aromatic. |
| D | 168.54 | 1 | For carbonyl carbon. |
| Total=18 | |||
Table 12: Carbon-13 NMR interpretations of compound 2c.
Conclusion
The authors referred to that new imatinib analogues have been happily designed, synthesized, and characterized in this research. The biological evaluation of these compounds is highly recommended.
Author Contributions
The authors are A.N. Hussein, O.F. Abdul-Rasheed, M.F. Mahdi, and A.M.R. Raauf. These authors contributed equally.
Conflicts of Interest
The authors confirmed that there are no conflicts of interest regarding publishing the current research.
Funding Sources
This research is self-funded.
REFERENCES
- Cheryl I, Christiansen S, Flanagin A. AMA Manual of Style: A Guide for Authors and Editors 10th. Oxfordshire: Oxford University Press. 2007.
- Meacham CE, Morrison SJ. Tumour heterogeneity and cancer cell plasticity. Nature. 2013;501:328-337.
- Ekins S, Mestres J, Testa B. In silico pharmacology for drug discovery: methods for virtual ligand screening and profiling. Br J Pharmacol. 2007;152:9-20.
- Qifeng B, Lanlan L, Shanhui L, Shujun X, Yu G. Drug Design Progress of In silico, In vitro and In vivo Researches. In-Vitro In-Vivo In-Silico J. 2018;1:16-37.
- Arora A, Scholar EM. Role of tyrosine kinase inhibitors in cancer therapy. J Pharmacol Exp Ther. 2005;315:971-979.
- Al-Obaidi ZMJ, Mousa TH, Ali AA. Synthesis of Novel Ibuprofen-Tranexamic Acid Codrug: Estimation of The Clinical Activity Against HCT116 Colorectal Carcinoma Cell Line and The Determination of Toxicity Profile Against MDCK Normal Kidney Cell Line. Int J Drug Deliv Technol. 2019;9:226-235.
- Al-Obaidi ZMJ, Rasheed OFA, Mahdi MF, Raauf AMR. In Silico Design, Synthesis and Characterization of New Spebrutinib Analogues. Pharm Anal Acta. 2019;10:612.
- https://patents.google.com/patent/ES2711249T3/en?q=heteroaryl&q=compounds&q=uses&oq=heteroaryl
- Evangelista Falcon W, Ellingson SR, Smith JC, Baudry J. Ensemble Docking in Drug Discovery: How Many Protein Configurations from Molecular Dynamics Simulations are Needed To Reproduce Known Ligand Binding?. J Phys Chem B. 2019;123:5189-5195.
- Lee YV, Choi SB, Wahab HA, Lim TS, Choong YS. Applications of Ensemble Docking in Potential Inhibitor Screening for Mycobacterium tuberculosis Isocitrate Lyase Using a Local Plant Database. J Chem Inf Model. 2019;59:2487-2495.
- Martínez-Fructuoso L, Pereda-Miranda R, Rosas-Ramírez D, Fragoso-Serrano M, Cerda-García-Rojas CM, Da Silva AS, et al. Structure elucidation, conformation, and configuration of cytotoxic 6-heptyl-5,6-dihydro-2 h -pyran-2-ones from hyptis species and their molecular docking to α-tubulin. J Nat Prod. 2019;82:520-531.
- Siebenmorgen T, Zacharias M. Evaluation of Predicted Protein-Protein Complexes by Binding Free Energy Simulations. J Chem Theory Comput. 2019;15:2071-2086.
- Bálint M, Horváth I, Mészáros N, Hetényi C. Towards Unraveling the Histone Code by Fragment Blind Docking. Int J Mol Sci. 2019;20:422.
- Jha P, Chaturvedi S, Swastika, Pal S, Jain N, Mishra AK. Improvising 5-HT 7 R homology model for design of high affinity ligands: model validation with docking, embrace minimization, MM-GBSA, and molecular dynamic simulations. J Biomol Struct Dyn. 2018;36:2475-2494.
- Lam PCH, Abagyan R, Totrov M. Ligand-biased ensemble receptor docking (LigBEnD): a hybrid ligand/receptor structure-based approach. J Comput Aided Mol Des. 2018;32:187-198.
- Padhorny D, Hall DR, Mirzaei H, Mamonov AB, Moghadasi M, Alekseenko A, et al. Protein-ligand docking using FFT based sampling: D3R case study. J Comput Aided Mol Des. 2018;32:225-230.
- Sekhar Pagadala N, Perez-Pineiro R, Bjorndahl TC, Wishart DS. Comparative molecular docking studies of EGCG with SHaPrPC. MOJ Drug Des Develop Ther. 2018;2:186-189.
- Pradeepkiran J, Reddy P. Structure Based Design and Molecular Docking Studies for Phosphorylated Tau Inhibitors in Alzheimer’s Disease. Cells. 2019;8:260.
- Śledź P, Caflisch A. Protein structure-based drug design: from docking to molecular dynamics. Curr Opin Struct Biol. 2018;48:93-102.
- Dean JA. Analytical Chemistry Handbook. New York: McGraw Hill, Inc. 1995;15.1–15.5. ISBN 0-07-016197-6.
- Lipinski CA, Lombardo F, Dominy BW, Feeney PJ. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv Drug Deliv Rev. 1997;46:3-25.
- Lipinski CA. Lead and drug-like compounds: the rule-of-five revolution. Drug Discovery Today: Technol. 2004;1:337-341.
- Oprea TI, Davis AM, Teague SJ, Leeson PD. Is there a difference between leads and drugs? A historical perspective. J Chem Inf Comput Sci. 2001;41:1308-1315.
- Leeson PD, Springthorpe B. The influence of drug-like concepts on decision-making in medicinal chemistry. Nat Rev Drug Discov. 2007;6:881-890.
- Li SK, Chantasart D. Skin Permeation Enhancement in Aqueous Solution: Correlation with Equilibrium Enhancer Concentration and Octanol/water Partition Coefficient. J Pharma Sci. 2019;108:350-357.
- Veber DF, Johnson SR, Cheng HY, Smith BR, Ward KW, Kopple KD. Molecular properties that influence the oral bioavailability of drug candidates. J med chem. 2002;45:2615-2623.
- Veber DF, Johnson SR, Cheng HY, Smith BR, Ward KW, Kopple KD. Molecular properties that influence the oral bioavailability of drug candidates. J med chem. 2002;45:2615-2623.
- Pajouhesh H, Lenz GR. Medicinal Chemical Properties of Successful Central Nervous System Drugs. NeuroRx. 2005;2:541–553.
- Hitchcock SA, Pennington LD. Structure Brain Exposure Relationships. J Med Chem. 2006;49:7559-7583.
- Caron G, Ermondi G. Molecular descriptors for polarity: the need for going beyond polar surface area. Future Med Chem. 2016;8:2013-2016.
- Vogel AI, Furniss BS, Tatchell AR, Hannaford AJ, Greig- Smith PW, Shankar R. Vogel's Textbook of Practical Organic Chemistry (5th Edition). Pearson. ISBN 978-0582462366.
- MCAT Organic Chemistry Review. Online + Book – Kaplan Test Prep. 2019.
- Brielle ES, Arkin IT. Site-Specific Hydrogen Exchange in a Membrane Environment Analyzed by Infrared Spectroscopy. J phys chem lett. 2018;9:4059-4065.
- Tan MJ, Pan HC, Tan HR, Chai JW, Lim QF, Wong TI et al. Flexible Modulation of CO‐Release Using Various Nuclearity of Metal Carbonyl Clusters on Graphene Oxide for Stroke Remediation. Adv Healthc Mater. 2018;7:1701113.
- Fadeeva VP, Tikhova VD, Nikulicheva ON. Elemental analysis of organic compounds with the use of automated CHNS analyzers. J Anal Chem. 2008;63:1094-1106.
- http://pubsapp.acs.org/paragonplus/submission/orlef7/orlef7_authguide.pdf?
- https://www.acs.org/content/acs/en/education/whatischemistry/landmarks/mri.html
- Dubey D, Kumar S, Chaurasia S, Guleria A, Ahmed S, Singh R, et al. NMR-Based Serum Metabolomics Revealed Distinctive Metabolic Patterns in Reactive Arthritis Compared with Rheumatoid Arthritis. J proteome res. 2018;18:130-146.
- Zhou B. The Applications of NMR Relaxometry, NMR Cryoporometry, and FFC NMR to Nanoporous Structures and Dynamics in Shale at Low Magnetic Fields. Energy & fuels. 2018;32:8897-8904.
- Puig-Castellví F, Pérez Y, Piña B, Tauler R, Alfonso I. Comparative analysis of 1H NMR and 1H–13C HSQC NMR metabolomics to understand the effects of medium composition in yeast growth. Anal chem. 2018;90:12422-12430.
- Yamada S, Ito K, Kurotani A, Yamada Y, Chikayama E, Kikuchi J. InterSpin: Integrated Supportive Webtools for Low-and High-Field NMR Analyses Toward Molecular Complexity. ACS Omega. 2019;4:3361-3369.
- Vyalikh A, Koroteev VO, Münchgesang W, Köhler T, Röder C, Brendler E, et al. Effect of Charge Transfer upon Li-and Na-Ion Insertion in Fine-Grained Graphitic Material as Probed by NMR. ACS Applied Materials & Interfaces. 2019;11:9291-9300.
- Sin M, Kavoosi N, Rauche M, Pallmann J, Paasch S, Senkovska I, et al. In Situ 13C NMR Spectroscopy Study of CO2/CH4 Mixture Adsorption by Metal–Organic Frameworks: Does Flexibility Influence Selectivity?. Langmuir. 2019;35:3162-3170.
- Carroll AW, Willis AC, Hoshino M, Kato A, Pyne SG. Corrected Structure of Natural Hyacinthacine C1 via Total Synthesis. J Nat Prod. 2019;82:358-367.
Citation: Hussein AN, Abdul-Rasheed OF, Mahdi MF, Raauf AMR (2019) Molecular Docking Studies for Design, Synthesis and Characterization of New Imatinib Analogues. Pharm Anal Acta 10:616. doi: 10.35248/2153-2435.19.10.616
Copyright: © 2019 Hussein AN, et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.