

Indexed In
- Open J Gate
- Genamics JournalSeek
- ResearchBible
- RefSeek
- Directory of Research Journal Indexing (DRJI)
- Hamdard University
- EBSCO A-Z
- OCLC- WorldCat
- Scholarsteer
- Publons
- MIAR
- Euro Pub
- Google Scholar
Useful Links
Share This Page
Journal Flyer

Open Access Journals
- Agri and Aquaculture
- Biochemistry
- Bioinformatics & Systems Biology
- Business & Management
- Chemistry
- Clinical Sciences
- Engineering
- Food & Nutrition
- General Science
- Genetics & Molecular Biology
- Immunology & Microbiology
- Medical Sciences
- Neuroscience & Psychology
- Nursing & Health Care
- Pharmaceutical Sciences
Research Article - (2023) Volume 12, Issue 1
Mitochondrial Genome Analysis and Phylogeny and Divergence Time Evaluation of the Strix aluco
David Smith1 and Renna Roy2*2Department of Environmental Sciences, American University, Washington, USA
Received: 24-Jan-2023, Manuscript No. BABCR-23-19645; Editor assigned: 27-Jan-2023, Pre QC No. BABCR-23-19645 (PQ); Reviewed: 10-Feb-2023, QC No. BABCR-23-19645; Revised: 17-Feb-2023, Manuscript No. BABCR-23-19645 (R); Published: 27-Feb-2023, DOI: 10.35248/2161-1009.23.12.472
Abstract
In this study, a complete mitochondrial genome of the Strix aluco was reported for the first time, with a total length of 18,632 bp. There were 37 genes, including 22 tRNAs, 2 rRNAs, 13 Protein-Coding Genes (PCGs), and 2 non-coding control regions (D-loop). The second-generation sequencing of the complete mitochondrial genome of the S. aluco was conducted using the Illumina platform, and then Tytoninae was used as the out-group, PhyloSuite software was applied to build the ML-tree and BI-tree of the Strigiformes, and finally, the divergence time tree was constructed using Beast 2.6.7 software, the age of Miosurniadiurnafossil-bearing sediments (6.0~9.5 mA) was set as the internal correction point. The common ancestor of the Strix was confirmed to have diverged during the Pleistocene (2.58~0.01 mA). The dramatic uplift of the Qinling Mountains in the Middle Pleistocene and the climate oscillation of the Pleistocene together caused Strix divergence between the northern and southern parts of mainland China. The isolation of glacial-interglacial rotation and glacier refuge was the main reason for the divergence of the common ancestor of the Strix uralensis and the S. aluco during this period. This study provides a reference for the evolution history of the Strix.
Keywords
Strix aluco; Phylogeny; Divergence time; Pleistocene; Climate oscillation; Mountains uplift
INTRODUCTION
Strix aluco belongs to Strigiformes, Strigidae, and is a medium-sized owl [1]. It is a non-migratory and territorial nocturnal bird [2,3]. It is widely distributed in the mountain broadleaf forest and mixed forest in Eurasia, and Israel is the southernmost country in the northern hemisphere [4,5]. Mammals, fish, amphibians, and even small birds such as sparrows can be its food, and voles are its most preferred food [6,7]. This species was listed as Least Concern (LC) on the International Union for Conservation of Nature (IUCN), and the current population trend is stable, the number of said individuals ranges from 1000000 to 2999999. The IUCN (2016): (https://www.iucnredlist.org/). In China, it has been listed as a national class II protected animal.
Mitochondria are characterized by maternal inheritance, high conservation, multiple copies in cells, low sequence recombination rate, and high evolutionary rate, widely used in phylogenetic studies and be able to accurately infer phylogenetic relationships in birds while complete mitochondrial genomes generally have higher accuracy than partial mitochondrial genes [8-12]. Previous studies have defined the phylogenetic position of S. aluco using a single gene or a combination of multiple mitochondrial genes [13-17]. Earlier studies identified the monophyly of Strigiformes phylogeny through the cytochrome B (Cyt B) gene through skeletal comparison, Striginae were divided into three subfamilies: Striginae (13 genera), Surniinae (8 genera) and Asioninae (2 genera) [18-19]. Phylogenetic relationships through the cytochrome B (Cyt B) gene also show that the Strigiformes can be divided into four parts, Tytoninae consists of Tytoninae (with Tyto) and Phodilinae (with Phodilus), and Striginae can be divided into Striginae, Surniinae and Ninoxinae. Among them, Striginae consists of six branches (Bubonini+Strigini+Pulsatrigini+Megascopini+Asionini+Otini), and Surniinae consists of three parts: Surnini+Athenini+Aegolini. Ninoxinae is mainly composed of Ninox, possibly including Sceloglaux completed the whole mitochondrial genome sequencing of Asio flammeus and determined the parallel phylogenetic relationship among the three genera of Otus, Ptilopsis and Asio; completed the whole mitochondrial genome sequencing of Strix uralensis, and determined the inter-genus relationship of Otus+ (Asio+ (Strix+Bubo)) through the study of the mitochondrial genome of Strigidae [20-23]. Clarified the global distribution of Tytonidae and the time of divergence, and their analysis showed that Tytonidae and S. aluco split from a common ancestor dating back to about 45 million years ago [24]. Identified the juxtaposition phylogenetic relationship between Striginaeand Surniinaein the South Asian Subcontinent population with Tytonidae as the out-group. Their study showed that Strigidaeand Tytonidaediverged at about 42.5~47.7 mA (mega-annum, million years). The timing of the divergence of the Strix is unclear.
There are many reasons for species divergence, among which geological and climatic influences on species diversification cannot be ignored [25]. The Cretacean-Tertiary extinction event was a mass extinction event in Earth’s history that occurred 65 million years ago and wiped out most of the animals and plants of the time, including the dinosaurs. It also wiped out the direct ancestors of tree-dwelling, water birds on Earth today; the few that survived evolved rapidly thereafter [26]. Bird ancestry began to increase exponentially at the end of the Eocene, from 100 species to 10,000 today [27]. Since the late Miocene, many birds in the Palaearctic have been migrating on a large scale and changing ranges have led to gene flows that have provided opportunities for the origin of various bird subfamilies [28,29]. Climatic oscillation during the Quaternary Period, especially throughout the Pleistocene (2.58~0.01 mA), promoted the evolution of species on a global scale, Pleistocene glacial gyre played a positive role in speciation [30-37] studied 14 populations of S. aluco in Western Europe and found that S. aluco in Europe could be divided into three branches originating from three glacial sanctuaries in the Iberian Peninsula, Italy and the Balkan Peninsula. This finding supports the “glacier refuge hypothesis” to describe the origin of S. aluco in Western Europe. The origin and divergence of S. aluco in mainland China are still a mystery.
There are many reasons for species divergence, among which geological and climatic influences on species diversification cannot be ignored [25]. The Cretacean-Tertiary extinction event was a mass extinction event in Earth’s history that occurred 65 million years ago and wiped out most of the animals and plants of the time, including the dinosaurs. It also wiped out the direct ancestors of tree-dwelling, water birds on Earth today; the few that survived evolved rapidly thereafter [26]. Bird ancestry began to increase exponentially at the end of the Eocene, from 100 species to 10,000 today [27]. Since the late Miocene, many birds in the Palaearctic have been migrating on a large scale and changing ranges have led to gene flows that have provided opportunities for the origin of various bird subfamilies [28,29]. Climatic oscillation during the Quaternary Period, especially throughout the Pleistocene (2.58~0.01 mA), promoted the evolution of species on a global scale, Pleistocene glacial gyre played a positive role in speciation [30-37] studied 14 populations of S. aluco in Western Europe and found that S. aluco in Europe could be divided into three branches originating from three glacial sanctuaries in the Iberian Peninsula, Italy and the Balkan Peninsula. This finding supports the “glacier refuge hypothesis” to describe the origin of S. aluco in Western Europe. The origin and divergence of S. aluco in mainland China are still a mystery.
Divergence time analysis can provide a reference for the evolution process of species and is also the basis for other further studies. In order to clarify the divergence time of species, it is necessary to obtain the gene sequence of species first, and then select an appropriate evolutionary model, and reliable calibration, such as the determining age of fossils [38, 39]. To clarify the phylogenetic position, divergence time and reasons of S. aluco from China, this study determined the complete mitochondrial genome of S. aluco, and used the mitochondrial genome combined with the mitochondrial genome of other birds in Strigiformes, The phylogenetic tree of Strigiformes was reconstructed. Fossil data are usually used to evaluate the divergence time of birds, and the divergence time of the Surniinae fossil is used as the correction point to analyse the divergence time of Strix, and the possible reasons for its divergence are fully discussed.
Materials and Methods
Sample origin and DNA extraction
Part of muscle tissue was extracted from the leg of a S. aluco that died of unknown cause in the Rescue Centre of Leigong Mountain National Nature Reserve, Qiandongnan Prefecture, Guizhou Province (26°49’26.40 “N, 104°43’ 33.60” E). Stored in refrigerated boxes with built-in thermometers, keeping the temperature near freezing. Transported back to the lab for DNA extraction. To extract DNA, we used the standardization CTAB method. The DNA Sample Prep Kit was used to construct genomic DNA libraries.
Sequencing and assembly
The Whole Genome Shotgun (WGS) strategy was used to construct the library [40]. The Next Generation Sequencing (NGS) technology was used for Paired-End sequencing (PE), based on the Illumina NovaSeq sequencing platform. The concentration and purity of DNA extracted from the samples were detected by Thermo Scientific Nano Drop 2000, and the integrity was detected by agarose electrophoresis and Agilent 2100 Bioanalyzer. Using the Covairs machine to break up DNA and fragment it. The gene library was constructed according to the shotgun method of Roe 2004. Agilent 2100 Bioanalyzer was used to detect the size of the library, and fluorescence quantitative detection was used to detect the total concentration of the library. The optimal amount of the library was selected and sequenced on Illumina. A single-stranded library was used as the template for bridge PCR amplification, and sequencing was performed while the synthesis.
After DNA extraction, purification, library construction and sequencing, the raw image file obtained by sequencing is the first, and the Raw Data that can be read in FASTQC format is generated after multi-step transformation, that is, the offline data. The data transformation work is automatically completed by the sequencing platform. According to the statistics of Raw data, 7,947,240 Reads (each sequence read is called one read) were obtained, the total number of bases was 1192,086000 bp, the percentage of fuzzy bases (uncertain bases) was 0.0016%, and the GC content was 44.58%. And base recognition accuracy of more than 99% accounted for 95.61% and base recognition accuracy of more than 99.9% accounted for 90.44%. The quality of the off-machine data should be tested through quality control, and the software used is FastQC (http://www.bioinformatics.babraham.ac.uk/projects/fastqc).
Sequencing data contains some low-quality reads with connectors, which will cause great interference to subsequent information analysis. In order to ensure the quality of subsequent information analysis, Fastp software (version 0.20.0) is needed to remove the contamination of sequencing connectors at the 3’ end. Low-quality sequences (sequences with an average Q value less than 20 and sequences with sequence length less than 50 bp) were removed; the number of high-quality reads obtained was 7611,984, accounting for 95.78% of the raw data, and the number of bases of high-quality reads was 1123739765 bp, accounting for 94.27% of the raw data [41].
A5-miseq v20150522 and SPAdesv3.9.0 were used for the de novo assembly of high-quality next-generation sequencing data. Construct contig and scaffold sequences. The sequences were extracted according to the sequencing depth of de novo splicing sequences, and the sequences with high sequencing depth were compared with the nt library on NCBI by blastn (BLAST v2.2.31+), and the mitochondrial sequences of each splicing result were selected. Integration of splicing results: The mitochondrial splicing results obtained by different software above were combined with reference sequences, and collinearity analysis was performed using mummer v3.1 software to determine the position relationship between contigs and fill gaps between contigs. The results were corrected using pilon v1.18 software to obtain the final mitochondrial sequence. The complete mitochondrial genome sequence obtained by splicing was uploaded to the MITOS web server (http://mitos2.bioinf.uni-leipzig.de/index.py) for functional annotation [42-46]. Among them, RefSeq 81 Metazoa is selected for Reference, The Genetic Code is set to a second set of vertebrate codons, and the rest are set according to the default parameters set by MITOS.
Through the above methods, the base composition of the whole mitochondrial genome, protein-coding genes, and rRNA genes was obtained. CGview visualization software was used to draw the mitochondrial complete genome circle map [47].
Mitochondrial genome data collection in Strigiformes
Currently, there are 30 species with mitochondrial genomes greater than 10000 bp in GenBank, including 27 species of Strigidae, and 3 species of Tytonidae, All taxonomic classifications of the species follow the current version of the IOC WORLD BIRD LIST (12.2) (http://dx.doi.org/10.14344/IOC.ML.12.2), the registration number as shown in (Table 1).
| Taxon | GenBank accession | Size (bp) | Notes | Reference |
|---|---|---|---|---|
| Aegolius funereus | MN122880 | 17166 | Partial | Direct Submission |
| Asio flammeus | KP889214 | 18966 | Complete | Zhang et al., 2004 |
| Asio otus | MG916810 | 17555 | Complete | Lee et al., 2018 |
| Athene brama | KF961185 | 16194 | Partial | Direct Submission |
| Athene noctua | MN122903 | 15776 | Partial | Direct Submission |
| Bubo blakistoni | LC099106 | 19379 | Partial | Direct Submission |
| Bubo bubo | MN206975 | 18956 | Complete | Direct Submission |
| Bubo flavipes | LC099100 | 19447 | Partial | Direct Submission |
| Bubo scandiacus | MG681084 | 18734 | Complete | Kang et al., 2018 |
| Ciccaba nigrolineata | MN356178 | 14875 | Partial | Feng et al., 2020 |
| Glaucidium brasilianum | MN356303 | 17717 | Partial | Feng et al., 2020 |
| Glaucidium brodiei | KP684122 | 17318 | Complete | Sun et al., 2016 |
| Glaucidium cuculoides | KY092431 | 17392 | Complete | Liu et al., 2019 |
| Ninox novaeseelandiae | AY309457 | 16223 | Complete | Harrison et al., 2004 |
| Ninox scutulata | KT943750 | 16208 | Complete | Direct Submission |
| Ninox strenua | KX529654 | 16206 | Complete | Sarker et al., 2016 |
| Otus bakkamoena | KT340631 | 17389 | Complete | Park et al., 2019a |
| Otus lettia | MW364567 | 16951 | Complete | Yu et al., 2021 |
| Otus scops | MW489467 | 17595 | Complete | Direct Submission |
| Otus semitorques | LC541473 | 18834 | Complete | Direct Submission |
| Otus sunia | KT340630 | 17413 | Complete | Park et al., 2019b |
| Sceloglaux albifacies | KX098448 | 15565 | Partial | Wood et al., 2017 |
| Strix aluco | MN122823 | 16490 | Partial | Feng et al., 2020 |
| Strix aluco | OP850567 | 18832 | Complete | This study |
| Strix leptogrammica | KC953095 | 16307 | Complete | Liu et al., 2014 |
| Strix occidentalis | MF431746 | 19889 | Complete | Hanna et al., 2017 |
| Strix uralensis | MG681081 | 18708 | Complete | Kang,H et al., 2018 |
| Strix varia Out group | MF431745 | 18975 | Complete | Hanna et al., 2017 |
| Phodilus badius | KF961183 | 17086 | Complete | Mahmood et al., 2014 |
| Tyto alba | EU410491 | 16148 | Partial | Pratt et al., 2009 |
| Tyto longimembris | KP893332 | 18466 | Partial | Xu et al., 2016 |
Table 1: Mitochondrial genome sequences used in this study.
Construction of phylogenetic trees
Using PhyloSuite software (download from: https://github.com/ dongzhang0725/PhyloSuite/releases), Drag the 30 GenBank format files downloaded from NCBI and the GenBank format files of S. aluco sequence obtained by this sequencing into the main interface.
First series of standardized operations, the choice for Mito genome sequence types, and then export the annotation error tRNA file, upload the ARWEN website (http://130.235.244.92/ARWEN/) modify comments, will be the site of the modified comments copy paste to modify after box. Secondly, 13 PCGs and 24 RNAs need to be extracted. The second set of 2 vertebrate mitochondrial codes is selected here, and the extracted 13 PCGs and 24 RNAs are imported into Multiple Alignment using Fast Fourier Transform (MAFFT) for multiple sequence alignment. Select the 37 gene files exported by MAFFT and import them into concatenate sequence, use the-auto strategy and normal alignment mode, and click start. Select the concatenated completion file and open the Partition Finder 2.0, performed a greedy search using the Bayesian, and calculated the optimal partitioning strategy and model selection. Using a separate GTR+G model for each data block [48].
Select the result file of Partition Finder 2.0 and complete the ML method in IQ-tree mode [49]. Set Phodilus badius, Tyto alba and Tyto longimembris as out-group. Under Edge-linked partition style for 10,000 replicates of ultrafast bootstrap also select the result folder of Partition Finder 2.0, open Mrbayes, set the out-groups, define parameters as Partition Models, run algebra as 2 parallel runs, 4 chains, 2,000,000 generations (must ensure the average standard deviation of split frequencies values were below 0.01), sampling freq is one sampling run for 1000 times, a burn-in number of initial 25% were burned [50,51].
Divergence time evaluation
Miosurnia diurna fossils provide an approximate date of the origin of Surniinae. The age of the fossil-bearing sediments of the M. diurna is 6.0-9.5 Am, Surniinae may be composed of the species of Surnia, Athene, Ninox, and Glaucidium, M.diurna fossil features are closer to the clade of Surnia+Glaucidium, Therefore, the origin times of Glaucidium brasilianum, Glaucidium brodiei, and Glaucidium cuculoides were set at 6 mA and 9.5 mA [52,53]. The ‘NEX’ file obtained by concatenating 37 genes using the concatenate sequence function in PhyloSuite was imported into BEAUti 2.6.7, (http://www.beast2.org/), Hasegawa–Kishino–Yano (HKY) model, with four gamma categories, Strict clock with 1.0 Clock rate, and with a Yule process (speciation) prior. Choose the Glaucidium brasilianum Glaucidium brodiei Glaucidium cuculoides (Sequence file name) to add Prior, Check the monophyletic option, and set Mean to 6.0/9.5 and Sigma to 0.1. A Markov Chain Monte Carlo (MCMC) Bayesian analysis with a chain length of 10 000 000, and with states recorded every 1000 iterations, save to run using BEAST 2.6.7. Log files were assessed using TRACER 1.7.2 (http://tree.bio.ed.ac.uk/software/tracer/) to ensure posteriors were normally distributed and all statistics had attained effective sample sizes of >200, if ESS<200, try to optimize by adding 5000000 iterations (chain length) each time. A burn-in of 10% was discarded, and a maximum clade credibility tree was determined and Mean heights were chosen using Tree Annotator 2.6.7. Finally, FigTree 1.4.4 was used to check the divergence time. Finally, use Adobe Illustrator 1.0.0.2 for visual editing.
Results and Discussion
Genome annotation
The total length of the mitochondrial genome sequence was 18,632 bp (Gen Bank entry number: OP850567). The genome annotation results showed that the total number of genes was 39, including 13 protein-coding genes, 22 tRNA genes, 2 rRNA genes, 2 OH genes, and 0 OL genes. Among them, 8 tRNA genes (trn-Q, trn-A, trn-N, trn-C, trn-Y, trn-P, trn-E, and trn-S2), one PCGs gene: nad6, are on the main chain (J chain); and the remaining 14 tRNA genes are trn-F, trn-V, trn-L2, trn-I, trn-M, trn-W, trn-D, trn-K, trn-G, trn-R, trn-H, trn-S1, trn-L1, trn-T; Two rRNA genes: rrn-S, rrn-L; And 12 PCGs genes encoding: nad1, nad2, nad3, nad4, nad4L, nad5, atp6, atp8, cox1, cox2, cox3, cytb on the secondary (N) chain (Table 2). There was no gene rearrangement (Figure 1). The specific annotation results of each gene are shown in Table 2.

Figure 1: Complete mitochondrial genome of S. aluco. The total length of the mitochondrial genome of S. aluco is 18632 Bp. The genes located on
the N strand or J strand are positioned inside or outside the circle. Contain two D-Loop regions. The GC Skew+region contains more Guanine
than Cytosine, and the GC Skew- region contains more Cytosine than Guanine. Note: 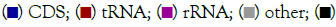 GC content;
GC content;  GC skew–.
GC skew–.
| Feature | Strand | Position | Length (bp) | Initiation Codon | Stop Codon | Anticodon | Intergenic Nucleotide |
|---|---|---|---|---|---|---|---|
| trnF | N | Jan-68 | 68 | - | - | GAA | -1 |
| rrnS | N | 68-1050 | 983 | - | - | - | -1 |
| trnV | N | 1050-1121 | 72 | - | - | TAC | 12 |
| rrnL | N | 1134-2709 | 1576 | - | - | - | -1 |
| trnL2 | N | 2709-2783 | 75 | - | - | TAA | 14 |
| nad1 | N | 2798-3757 | 960 | ATG | AGG | - | -2 |
| trnI | N | 3756-3827 | 72 | - | - | GAT | 11 |
| trnQ | J | 3839-3909 | 71 | - | - | TTG | -1 |
| trnM | N | 3909-3977 | 69 | - | - | CAT | - |
| nad2 | N | 3978-5018 | 1041 | ATG | TAG | - | -2 |
| trnW | N | 5017-5091 | 75 | - | - | TCA | 1 |
| trnA | J | 5093-5161 | 69 | - | - | TGC | 1 |
| trnN | J | 5163-5236 | 74 | - | - | GTT | 2 |
| trnC | J | 5239-5305 | 67 | - | - | GCA | -1 |
| trnY | J | 5305-5376 | 72 | - | - | GTA | 1 |
| cox1 | N | 5378-6928 | 1551 | ATG | AGG | - | -9 |
| trnS2 | J | 6920-6991 | 72 | - | - | TGA | 3 |
| trnD | N | 6995-7063 | 69 | - | - | GTC | 2 |
| cox2 | N | 7066-7749 | 684 | ATG | TAA | - | 9 |
| trnK | N | 7759-7828 | 70 | - | - | TTT | 1 |
| atp8 | N | 7830-7997 | 168 | ATG | TAA | - | -10 |
| atp6 | N | 7988-8671 | 684 | ATG | TAA | - | -1 |
| cox3 | N | 8671-9454 | 784 | ATG | T(AA) | - | - |
| trnG | N | 9455-9523 | 69 | - | - | TCC | - |
| nad3 | N | 9524-9875 | 352 | ATG | TAA | - | 2 |
| trnR | N | 9878-9946 | 69 | - | - | TCG | 1 |
| nad4l | N | 9948-10244 | 297 | ATG | TAA | - | -7 |
| nad4 | N | 10238-11615 | 1378 | ATG | T(AA) | - | - |
| trnH | N | 11616-11685 | 70 | - | - | GTG | 2 |
| trnS1 | N | 11688-11753 | 66 | - | - | GCT | 2 |
| trnL1 | N | 11756-11826 | 71 | - | - | TAG | -15 |
| nad5 | N | 11812-13647 | 1836 | ATG | TAA | - | 5 |
| cob | N | 13653-14795 | 1143 | ATG | TAA | - | 1 |
| trnT | N | 14797-14866 | 70 | - | - | TGT | 728 |
| OH_0 | N | 15595-15792 | 198 | - | - | - | 616 |
| trnP | J | 16409-16478 | 70 | - | - | TGG | 6 |
| nad6 | J | 16485-17006 | 522 | ATG | TAG | - | 3 |
| trnE | J | 17010-17083 | 74 | - | - | TTC | 584 |
| OH_1 | N | 17668-17865 | 198 | - | - | - | 766 |
Table 2: Analysis of mitochondrial genome feature.
Phylogenetic analysis
In this study, both ML-tree and BI-tree show the same tree topology with good support, and it can be seen from the tree that Strigidae and Tytonidae are two distinct lineages under the owl shape. Ciccaba nigrolineata is nested in the Strix, and Sceloglaux albifacies is nested in the genus Ninox. S. aluco in this study is a sister group of S. uralensis, Strix aluco MN122823+ (Strix aluco OP850567+Strix uralensis) was formed with S. aluco; Athene noctua is a sister group of Athene brama, Aegolius funereus is a sister group of Glaucidium cuculoides, Glaucidium brasilianum; Glaucidium brodiei, G. cuculoides, G. brasilianum, Athene noctua, A. brama, A. funereus constitute the same group; Strix and Bubo are a sister group. It forms an Asio+ (Strix+Bubo) monophyletic group with Asio; And a higher monophyletic group with Otus+(Asio+ (Strix+Bubo)), this monophyly simultaneously with (Sceloglaux albifacies+Ninox) monophyly exhibited as dyadic taxa (Figure 2).

Figure 2: BI/ML-tree-Bayesian phylogenetic tree of 37 genes (24 rRNAs, 13PCGs) from 31 species of Strigiformes. The node labels are BI/ML posterior probability and bootstrap support value respectively, and the scale indicates the probability of nucleotide change within each branch length. The GenBank of the sequences has been indicated next to the species name. Branches of different subfamilies are distinguished by different colors, with Tytoninae (with Phodilus badius, Tyto longimembris and Tyto alba) being the out-group. The S. aluco mitochondrial genome obtained by this sequencing has been marked by ★
Divergence time evaluation
The divergence time tree based on 37 genomes shows that the time interval between Strigidaeand Tytonidaefrom the common ancestor of Strigiformeswas 8.69~13.76 mA. However, in the out-group, Tytoalba, Tytolongimembrisand Phodilusbadiusdiverged from the common ancestor about 4.57~7.24 mA. The divergence of Strigidae began about 6.81-10.79 mA, and the earliest divergence of Surniinae occurred in Strigidae, and Aegolius, Glaucidium, and Athene occurred about 6.03-9.55 mA.
In this study, we found the divergence time between S. aluco (OP850567) and S.alucoof Margaryan. A (MN122823) was about 1.59~2.51 mA, and the divergence time between S.aluco and S. uralensis in China was about 1.38~2.19 mA (Figure 3).

Figure 3: Divergence time tree. Through the divergence time tree obtained by Beast 2.6.7 based 724 on the Bayesian method, the node horizontal bar indicates that the posterior probability of this age 725 interval is 95%, and the divergence time has been marked at the node. The S. aluco mitochondrial genome obtained by this sequencing has been marked by ★
The mitochondrial genome structure of birds is a covalent double-chain loop structure, with a total of 37 genes, including 22 tRNAs, 2 rRNAs, 13 Protein-Coding Genes (PCGs) and 1-2 non-coding control regions (D-loop). Where nad6 and 8 tRNA encoding genes (trnQ, trnA, trnN, trnC, trnY, trnS2, trnP and trnE) are on the J chain (light chain), The remaining 14 tRNAs, 2 rRNAs, 12 protein- coding genes and 1-2 non-coding control regions (D-loop) are all on the N chain (heavy chain), and the complete mitochondrial genome structure of all birds was consistent [54-56]. The complete mitochondrial genome sequence of S. aluco obtained in this study was circular, with a total length of 18,632 bp and a GC content of 46.76%. Its composition was as follows: the proportion of Adenine bases in the total base column (A%) was 29.57%; The ratio of Guanine base to total base (G%) was 14.09%. The ratio of Cytosine base to total base (C%) was 32.67%. The ratio of Thymidine to the total base column (T%) was 23.66%. The start codon of all 13 PCGs is ATG, and the transcription stop codon is AGG, TAG and TAA. The content of A+T (53.23%) was higher than that of G+C (46.76%), which was consistent with the AT tendency of base bias in the vertebrate mitochondrial genome, which is also consistent with the mitochondrial genome of other owls in Strigidae [57-60].
Phylogenetic analysis of S. aluco
The BI and the ML tree have a consistent topology and each node has a high posterior probability. The phylogenetic tree of Strigiformes obtained by the mitochondrial genome in this study is consistent with the phylogenetic tree obtained by through morphology. Our phylogenetic tree shows that compared Surnini (with Surnia, Glaucidium and Taenioglaux), Athenini (with Athene) and Aegolini (with Aegolius) under the Surniinae are feasible. In the phylogenetic tree constructed by, C. nigrolineata was also nested in Strix. S. albifacies has been largely extinct on the island of New Zealand, extracted its mitochondrial genome from museum specimens and suggested changing its name to Ninox albifacies because it has the same morphological structure and phylogenetic position as the genus Ninox. S. aluco in this study is a sister group of S. uralensis uploaded to GenBank by [61-65]. While S. aluco uploaded with Margaryan. A form a monophyly of Strix aluco MN122823+ (Strix aluco OP850567+Strix uralensis). Compared with the mitochondrial genome of S. aluco obtained by Margaryan. A, S. aluco in China is more closely related to the S. uralensis, probably because at the beginning of the Pleistocene, the common ancestor of S. aluco MN122823 and S. aluco OP850567 had already been geographically isolated. The isolation of the Pleistocene refugium led to the divergence of the whole genome of the common ancestor of the forest owl at home and abroad. Foreign studies have shown that the Quaternary Period is characterized by a series of glacial-interglacial cycles, with the ancestors of modern species seeking refuge in a suitable environment. The existing species on the Qinghai-Tibet Plateau may be the result of rapid population expansion in relatively war refugia during the Pleistocene glaciation and interglacial period, forming the current distribution pattern and genetic diversity [66,67]. Legong Mountain in Guizhou Province just played the role of refugia for S. aluco during the Pleistocene glaciation period. Mitochondrial phylogeographic studies show that the origin of S. alucoin Western Europe supports the glacial refuge hypothesis, and that the species survived in three allopatric refugees in the Iberian Peninsula, Italy and the Balkans, becoming the main source of S. aluco in Europe during the late glacial period. DNA barcoding technology also proved that the geographical barrier of the Strait of Gibraltar played an extremely important role in the phylogenetic history of S. aluco [68,69]. The phylogenetic relationship of Strigidae forms a phylogenetic relationship of Surniinae+(Ninox+(Otus+ (Asio+ (Strix+Bubo)))), The conclusion of Otus+ (Asio+ (Strix+Bubo)) is consistent with the conclusion of Kang et al. (2018). According to the genome analysis of Strigidae birds in Madagascar, Strix is most closely related to Bubo, followed by Otus [70].
Divergence time evaluation of S. aluco
On the Qinghai-Tibet Plateau, the impact of mountain uplift on the formation of modern species (<2.0 mA) is limited, and researchers are more willing to believe that climate fluctuations played a key role in the formation of species in the Middle Pleistocene [71,72]. During the Quaternary Period and Pleistocene (1.6~2.7 mA), there were severe climate shocks, which played a positive role in promoting the formation of species [73-76]. The Pleistocene began 2.58 million years ago (2.58 mA). Climate fluctuations during this period, especially during the ice age, affected the distribution of forests in the Northern Hemisphere and the evolution of species living in forests [77]. This series of climate fluctuations in the Pleistocene promoted species variation. This has led to species differentiation [78]. Glaciation has played an important role in influencing the population size, species and community genetic structure of today’s species, the glacial-interglacial gyrations of the same period also affected the distribution of species, Glacial- interglacial cycles led to periodic shifts in glacial refuges for Pleistocene birds, the isolation of glacier refugia will lead to the divergence of the whole genome of species, thus forming different species, this should be the reason why the common ancestor of S. aluco MN122823 and S. aluco OP850567 (this study) diverged at 1.59~2.51 mA, genetic divergence of the same lineage due to the isolation of refugees leads to lineage divergence. All kinds of species generally begin to migrate to the best habitat during the warm climate period, in particular, species adapted to low altitudes in the early stage of climate change will move to high altitudes at this time, resulting in the reproductive isolation of species in the two places [80-89]. During the Pleistocene-Holocene (1.10~0.60 mA), the Qinghai-Tibet Plateau experienced three stages of rapid uplift, with mountains forming, climate-changing from moist and warm to dry and cold, and forests retreating to the edge of the plateau [90]. The Quaternary Period climate shock led to the initial formation of the existing forest and mountain distribution pattern in the Northern Hemisphere. Birds began to be widely distributed after leaving the glacier refuge at the end of the glacier and initially formed the existing distribution pattern [91]. The long-tailed forest owl may have moved north at this time and thus diverged from S. aluco. In addition, the rapid uplift of the Qinling Mountains from the end of the Early Pleistocene to the Middle Pleistocene may have made the Qinling Mountains a barrier to north-south bird communication [92].
Conclusion
The rapid uplift of the Qinling Mountains prevented communication between S. aluco and the common ancestor of S.uralensis, which was originally distributed on the north and south sides. In summary, by sequencing the complete mitochondrial genome of S. aluco, and mapping its phylogenetic tree and divergence time tree, the phylogenetic relationship of Strigiformes(Tytoninae+Phodilinae)+(Striginae+Ninoxinae+Surniinae) is summarized, with Tytonidae including Tytoninae (with Tyto) and Phodilinae (with Phodilus) as the out-group, Strigidae comprises Striginae (with Asio, Bubo, Strix, Ciccaba and Otus)+Ninoxinae+Surniinae (with Athenini, Aegoliniand Glaucidium). The divergence time tree showed that the divergence time between S. aluco of China and S.aluco of other countries was about 1.59~2.51 mA, suggesting that the common ancestor of S. aluco at home and abroad was separated by geographical isolation at the beginning of the Pleistocene. The divergence between S. aluco and S. uralensis in China was about 1.38~2.19 mA. During this time, the rapid uplift of the Qinling Mountains led to the divergence of the ancestors of Strix on the north and south sides of the Chinese mainland. At the same time, due to Climatic oscillation in the Pleistocene, the existing S. aluco population on the Qinghai-Tibet Plateau may have rapidly expanded in relatively warm shelters such as Leigong Mountain to form the current distribution pattern.
Summary Statement
This study was the first time to assemble the complete mitochondrial genome of Strix aluco, and report the divergence time of Strix. A full discussion was made, and it was inferred that the uplift of the Qinling Mountains and the glacial refuge led to the differentiation of this genus.
Data Availability
The complete mitochondrial genome of S. aluco has been uploaded to NCBI, GenBank accession number: OP850567.
Conflict of Interest
The authors declare no competing or financial interests.
Funding
Guizhou Provincial Science and Technology Foundation, Grant/ Award Number: Qiankehe LH (2020) 1Y080; Project supported by the Joint Fund of the National Natural Science Foundation of China and the Karst Science Research Center of Guizhou province, Grant/Award Number: U1812401; Science and Technology Foundation of Guizhou Forestry Bureau (Qianlinkehe (2020) 09), and Guizhou Universtiy Dr. Scientific Research Fund (Guidarenjihe (2018) 07).
Acknowledgement
Thank Rescue Center of Leigong Mountain National Nature Reserve, Qiandongnan Prefecture, Guizhou Province (26°49’26.40 “N, 104°43’ 33.60” E) for provides the tissue slice of strix aluco.
References
- Grytsyshina EE, Kuznetsov AN, Panyutina AA. Kinematic constituents of the extreme head turn of Strix aluco estimated by means of CT-scanning. Dokl Biol Sci. 2016; 466:(24-27). Pleiades Publishing.
[Crossref] [Google Scholar] [PubMed]
- Doña J, Ruiz-Ruano FJ, Jovani R. DNA barcoding of Iberian Peninsula and North Africa Tawny Owls Strix aluco suggests the Strait of Gibraltar as an important barrier for phylogeography. Mitochondrial DNA A DNA Mapp Seq Anal. 2016; 27(6):4475-4478.
[Crossref] [Google Scholar] [PubMed]
- Sunde P, Bølstad MS, Desfor KB. Diurnal exposure as a risk sensitive behaviour in tawny owls Strix aluco? J Avian Biol. 2003; 34(4):409-418.
- Obuch J. Spatial and temporal diversity of the diet of the tawny owl (Strix aluco).2011.
- Comay O, Ezov E, Yom-Tov Y, Dayan T. In its southern edge of distribution, the tawny owl (strix aluco) is more sensitive to extreme temperatures than to rural development. Animals (Basel). 2022;12(5):641.
[Crossref] [Google Scholar] [PubMed]
- Solonen T, Karhunen J. Effects of variable feeding conditions on the Tawny Owl Strix aluco near the northern limit of its range. Ornis Fennica. 2002; 79(3):121-131.
- Karell P, Ahola K, Karstinen T, Zolei A, Brommer JE. Population dynamics in a cyclic environment: consequences of cyclic food abundance on tawny owl reproduction and survival. J Anim Ecol. 2009; 78(5):1050-1062.
[Crossref] [Google Scholar] [PubMed]
- Yan C, Mou B, Meng Y, Tu F, Fan Z, Price M, et al. A novel mitochondrial genome of Arborophila and new insight into Arborophila evolutionary history. PLoS One. 2017;12(7):e0181649.
[Crossref] [Google Scholar] [PubMed]
- Sun CH, Liu HY, Min X, Lu CH. Mitogenome of the little owl Athene noctua and phylogenetic analysis of Strigidae. Int J Biol Macromol. 2020; 151:924-931.
[Crossref] [Google Scholar] [PubMed]
- Tuinen MV, Sibley CG, Hedges SB. The early history of modern birds inferred from DNA sequences of nuclear and mitochondrial ribosomal genes. Mol Biol Evol. 2000; 17(3):451-457.
[Crossref] [Google Scholar] [PubMed]
- Haring E, Kruckenhauser L, Gamauf A, Riesing MJ, Pinsker W. The complete sequence of the mitochondrial genome of Buteo buteo (Aves, Accipitridae) indicates an early split in the phylogeny of raptors. Mol Biol Evol. 2001; 18(10):1892-1904.
[Crossref] [Google Scholar] [PubMed]
- Harrison GL, McLenachan PA, Phillips MJ, Slack KE, Cooper A, Penny D, et al. Four new avian mitochondrial genomes help get to basic evolutionary questions in the late cretaceous. Mol Biol Evol. 2004; 21(6):974-983.
[Crossref] [Google Scholar] [PubMed]
- Heidrich P, Wink M. Tawny owl (Strix aluco) and Hume's tawny owl (Strix butleri) are distinct species: evidence from nucleotide sequences of the cytochrome b gene. Z Naturforsch C J Biosci. 1994; 49(3-4):230-234.
[Crossref] [Google Scholar] [PubMed]
- Yu J, Liu J, Li C, Wu W, Feng F, Wang Q, et al. Characterization of the complete mitochondrial genome of Otus lettia: exploring the mitochondrial evolution and phylogeny of owls (Strigiformes). Mitochondrial DNA B Resour. 2021; 6(12):3443-3451.
[Crossref] [Google Scholar] [PubMed]
- Li Z, Stidham TA, Zheng X, Wang Y, Zhao T, Deng T, et al. Early evolution of diurnal habits in owls (Aves, Strigiformes) documented by a new and exquisitely preserved Miocene owl fossil from China. Proc Natl Acad Sci U S A. 2022; 119(15):e2119217119.
[Crossref] [Google Scholar] [PubMed]
- Fuchs J, Pons JM, Goodman SM, Bretagnolle V, Melo M, Bowie RC, et al. Tracing the colonization history of the Indian Ocean scops-owls (Strigiformes: Otus) with further insight into the spatio-temporal origin of the Malagasy avifauna. BMC Evol Biol.2008; 8(1):1-5.
[Crossref] [Google Scholar] [PubMed]
- Wood JR, Mitchell KJ, Scofield RP, De Pietri VL, Rawlence NJ, Cooper A, et al. Phylogenetic relationships and terrestrial adaptations of the extinct laughing owl, Sceloglaux albifacies (Aves: Strigidae). Zool J Linn Soc. 2017;179(4):907-918.
- Wink M, Heidrich P. Molecular systematics of owls (Strigiformes) based on DNA-sequences of the mitochondrial cytochrome b gene.2000.
- Salter JF, Oliveros CH, Hosner PA, Manthey JD, Robbins MB, Moyle RG, et al. Extensive paraphyly in the typical owl family (Strigidae). The Auk. 137(1):ukz070.
- Wink M, El-Sayed AA, Sauer-Gürth H, Gonzalez J. Molecular phylogeny of owls (Strigiformes) inferred from DNA sequences of the mitochondrial cytochrome b and the nuclear RAG-1 gene. Ardea. 2009; 97(4):581-591.
- Zhang Y, Song T, Pan T, Sun X, Sun Z, Qian L, et al. Complete sequence and gene organization of the mitochondrial genome of Asio flammeus (Strigiformes, strigidae). Mitochondrial DNA A DNA Mapp Seq Anal. 2016;27(4):2665-2667.
[Crossref] [Google Scholar] [PubMed]
- Kang H, Li B, Ma X, Xu Y. Evolutionary progression of mitochondrial gene rearrangements and phylogenetic relationships in Strigidae (Strigiformes). Gene. 2018; 674:8-14.
[Crossref] [Google Scholar] [PubMed]
- Uva V, Päckert M, Cibois A, Fumagalli L, Roulin A. Comprehensive molecular phylogeny of barn owls and relatives (Family: Tytonidae), and their six major Pleistocene radiations. Mol Phylogenet Evol. 2018;125:127-137.
[Crossref] [Google Scholar] [PubMed]
- Koparde P, Mehta P, Reddy S, Ramakrishnan U, Mukherjee S, Robin VV, et al. The critically endangered forest owlet Heteroglaux blewitti is nested within the currently recognized Athene clade: A century-old debate addressed. PLoS One. 2018; 13(2):e0192359.
[Crossref] [Google Scholar] [PubMed]
- Claramunt S, Cracraft J. A new time tree reveals Earth history’s imprint on the evolution of modern birds. Sci Adv. 2015; 1(11):e1501005.
[Crossref] [Google Scholar] [PubMed]
- Field DJ, Bercovici A, Berv JS, Dunn R, Fastovsky DE, Lyson TR, et al. Early evolution of modern birds structured by global forest collapse at the end-Cretaceous mass extinction. Curr Biol. 2018; 28(11):1825-1831.
[Crossref] [Google Scholar] [PubMed]
- Ksepka DT, Phillips MJ. Avian Diversification Patterns across the K-Pg Boundary: Influence of Calibrations, Datasets, and Model Misspecification. Ann Missouri Bot Gard. 2015; 100(4):300-328.
- Drovetski SV. Plio‐Pleistocene climatic oscilations, Holarctic biogeography and speciation in an avian subfamily. J Biogeogr. 2003; 30(8):1173-1181.
- Holm SR, Svenning JC. 180,000 years of climate change in Europe: avifaunal responses and vegetation implications. PLoS One. 2014; 9(4):e94021.
[Crossref] [Google Scholar] [PubMed]
- Hewitt G. The genetic legacy of the Quaternary ice ages. Nature. 2000;405(6789):907-913.
[Crossref] [Google Scholar] [PubMed]
- Hewitt GM. Genetic consequences of climatic oscillations in the Quaternary. Philos Trans R Soc Lond B Biol Sci. 2004; 359(1442):183-195.
[Crossref] [Google Scholar] [PubMed]
- Lamb AM, Gonçalves da Silva A, Joseph L, Sunnucks P, Pavlova A. Pleistocene-dated biogeographic barriers drove divergence within the Australo-Papuan region in a sex-specific manner: an example in a widespread Australian songbird. Heredity. 2019; 123(5):608-621.
[Crossref] [Google Scholar] [PubMed]
- Kozma R, Lillie M, Benito BM, Svenning JC, Höglund J. Past and potential future population dynamics of three grouse species using ecological and whole genome coalescent modeling. Ecol Evol. 2018; 8(13):6671-6681.
[Crossref] [Google Scholar] [PubMed]
- Zhao S, Zheng P, Dong S, Zhan X, Wu QI, Guo X, et al. Whole-genome sequencing of giant pandas provides insights into demographic history and local adaptation. Nat Genet. 2013;45(1):67-71.
[Crossref] [Google Scholar] [PubMed]
- Hung CM, Shaner PJ, Zink RM, Liu WC, Chu TC, Huang WS, et al. Drastic population fluctuations explain the rapid extinction of the passenger pigeon. Proc Natl Acad Sci U S A. 2014; 111(29):10636-10641.
[Crossref] [Google Scholar] [PubMed]
- Mays HL, Hung CM, Shaner PJ, Denvir J, Justice M, Yang SF, et al. Genomic analysis of demographic history and ecological niche modeling in the endangered Sumatran rhinoceros Dicerorhinus sumatrensis. Curr Biol. 2018; 28(1):70-76.
[Crossref] [Google Scholar] [PubMed]
- Brito PH. The influence of Pleistocene glacial refugia on tawny owl genetic diversity and phylogeography in western Europe. Mol Ecol. 2005; 14(10):3077-3094.
[Crossref] [Google Scholar] [PubMed]
- Ho SY, Duchêne S. Molecular‐clock methods for estimating evolutionary rates and timescales. Mol Ecol. 2014; 23(24):5947-5965.
[Crossref] [Google Scholar] [PubMed]
- Ho SY, Phillips MJ. Accounting for calibration uncertainty in phylogenetic estimation of evolutionary divergence times. Syst Biol. 2009; 58(3):367-380.
[Crossref] [Google Scholar] [PubMed]
- Roe BA. Shotgun library construction for DNA sequencing. Methods Mol Bio. 2004; 255:171-187.
[Crossref] [Google Scholar] [PubMed]
- Chen S, Zhou Y, Chen Y, Gu J. fastp: an ultra-fast all-in-one FASTQ preprocessor. Bioinformatics. 2018; 34(17):i884-i900.
[Crossref] [Google Scholar] [PubMed]
- Coil D, Jospin G, Darling AE. A5-miseq: an updated pipeline to assemble microbial genomes from Illumina MiSeq data. Bioinformatics. 2015;31(4):587-589.
[Crossref] [Google Scholar] [PubMed]
- Bankevich A, Nurk S, Antipov D, Gurevich AA, Dvorkin M, Kulikov AS,et al. SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J Comput Biol. 2012 ;19(5):455-477.
[Crossref] [Google Scholar] [PubMed]
- Kurtz S, Phillippy A, Delcher AL, Smoot M, Shumway M, Antonescu C, et al. Versatile and open software for comparing large genomes. Genome Biol. 2004; 5:1-9.
[Crossref] [Google Scholar] [PubMed]
- Walker BJ, Abeel T, Shea T, Priest M, Abouelliel A, Sakthikumar S, et al. Pilon: an integrated tool for comprehensive microbial variant detection and genome assembly improvement. PLoS One. 2014;9(11):e112963.
[Crossref] [Google Scholar] [PubMed]
- Bernt M, Donath A, Jühling F, Externbrink F, Florentz C, Fritzsch G,et al. MITOS: improved de novo metazoan mitochondrial genome annotation. Mol Phylogenet Evol. 2013; 69(2):313-319.
[Crossref] [Google Scholar] [PubMed]
- Stothard P, Wishart DS. Circular genome visualization and exploration using CGView. Bioinformatics. 2005;21(4):537-539.
[Crossref] [Google Scholar] [PubMed]
- Lanfear R, Frandsen PB, Wright AM, Senfeld T, Calcott B. PartitionFinder 2: new methods for selecting partitioned models of evolution for molecular and morphological phylogenetic analyses. Mol Biol Evol. 2017; 34(3):772-773.
[Crossref] [Google Scholar] [PubMed]
- Minh BQ, Schmidt HA, Chernomor O, Schrempf D, Woodhams MD, von Haeseler A, et al. Corrigendum to: IQ-TREE 2: new models and efficient methods for phylogenetic inference in the genomic era. Mol Biol Evol.2020;37(8):2461.
[Crossref] [Google Scholar] [PubMed]
- Hoang DT, Chernomor O, Von Haeseler A, Minh BQ, Vinh LS. UFBoot2: improving the ultrafast bootstrap approximation. Mol Biol Evol. 2018; 35(2):518-522.
[Crossref] [Google Scholar] [PubMed]
- Sterli J, Pol D, Laurin M. Incorporating phylogenetic uncertainty on phylogeny‐based palaeontological dating and the timing of turtle diversification. Cladistics. 2013; 29(3):233-246.
[Crossref] [Google Scholar] [PubMed]
- Li Z, Stidham TA, Zheng X, Wang Y, Zhao T, Deng T, et al. Early evolution of diurnal habits in owls (Aves, Strigiformes) documented by a new and exquisitely preserved Miocene owl fossil from China. Proc Natl Acad Sci U S A. 2022; 119(15):e2119217119.
[Crossref] [Google Scholar] [PubMed]
- Wink M, El-Sayed AA, Sauer-Gürth H, Gonzalez J. Molecular phylogeny of owls (Strigiformes) inferred from DNA sequences of the mitochondrial cytochrome b and the nuclear RAG-1 gene. Ardea. 2009; 97(4):581-591.
- Wolstenholme DR. Animal mitochondrial DNA: structure and evolution. Int Rev Cytol. 1992;141:173-216.
[Crossref] [Google Scholar] [PubMed]
- Boore JL. Animal mitochondrial genomes. Nucleic Acids Res. 1999; 27(8):1767-1780.
[Crossref] [Google Scholar] [PubMed]
- Hanna ZR, Henderson JB, Sellas AB, Fuchs J, Bowie RC, Dumbacher JP, et al. Complete mitochondrial genome sequences of the northern spotted owl (Strix occidentalis caurina) and the barred owl (Strix varia; Aves: Strigiformes: Strigidae) confirm the presence of a duplicated control region. PeerJ. 2017 ;5:e3901.
[Crossref] [Google Scholar] [PubMed]
- Broughton RE, Milam JE, Roe BA. The complete sequence of the zebrafish (Danio rerio) mitochondrial genome and evolutionary patterns in vertebrate mitochondrial DNA. Genome Res. 2001; 11(11):1958-1967.
[Crossref] [Google Scholar] [PubMed]
- Ma Z, Yang X, Bercsenyi M, Wu J, Yu Y, Wei K, et al. Comparative mitogenomics of the genus Odontobutis (Perciformes: Gobioidei: Odontobutidae) revealed conserved gene rearrangement and high sequence variations. Int J Mol Sci. 2015; 16(10):25031-25049.
[Crossref] [Google Scholar] [PubMed]
- Sun CH, Liu HY, Min X, Lu CH. Mitogenome of the little owl Athene noctua and phylogenetic analysis of Strigidae. Int J Biol Macromol. 2020; 151:924-931.
[Crossref] [Google Scholar] [PubMed]
- Kang H, Li B, Ma X, Xu Y. Evolutionary progression of mitochondrial gene rearrangements and phylogenetic relationships in Strigidae (Strigiformes). Gene. 2018; 674:8-14.
[Crossref] [Google Scholar] [PubMed]
- Li Z, Stidham TA, Zheng X, Wang Y, Zhao T, Deng T, et al. Early evolution of diurnal habits in owls (Aves, Strigiformes) documented by a new and exquisitely preserved Miocene owl fossil from China. Proc Natl Acad Sci U S A. 2022; 119(15):e2119217119.
[Crossref] [Google Scholar] [PubMed]
- Wink M, El-Sayed AA, Sauer-Gürth H, Gonzalez J. Molecular phylogeny of owls (Strigiformes) inferred from DNA sequences of the mitochondrial cytochrome b and the nuclear RAG-1 gene. Ardea. 2009; 97(4):581-591.
- Yu J, Liu J, Li C, Wu W, Feng F, Wang Q, et al. Characterization of the complete mitochondrial genome of Otus lettia: exploring the mitochondrial evolution and phylogeny of owls (Strigiformes). Mitochondrial DNA B Resour. 2021; 6(12):3443-3451.
[Crossref] [Google Scholar] [PubMed]
- Wood JR, Mitchell KJ, Scofield RP, De Pietri VL, Rawlence NJ, Cooper A, et al. Phylogenetic relationships and terrestrial adaptations of the extinct laughing owl, Sceloglaux albifacies (Aves: Strigidae). Zool J Linn Soc. 2017;179(4):907-918.
- Kang H, Li B, Ma X, Xu Y. Evolutionary progression of mitochondrial gene rearrangements and phylogenetic relationships in Strigidae (Strigiformes). Gene. 2018; 674:8-14.
[Crossref] [Google Scholar] [PubMed]
- Woodruff DS. Biogeography and conservation in Southeast Asia: how 2.7 million years of repeated environmental fluctuations affect today’s patterns and the future of the remaining refugial-phase biodiversity. Biodivers Conserv. 2010; 19:919-941.
- Gao YD, Zhang Y, Gao XF, Zhu ZM. Pleistocene glaciations, demographic expansion and subsequent isolation promoted morphological heterogeneity: A phylogeographic study of the alpine Rosa sericea complex (Rosaceae). Sci Rep. 2015; 5(1):1-5.
[Crossref] [Google Scholar] [PubMed]
- Brito PH. The influence of Pleistocene glacial refugia on tawny owl genetic diversity and phylogeography in western Europe. Mol Ecol. 2005; 14(10):3077-3094.
[Crossref] [Google Scholar] [PubMed]
- Doña J, Ruiz-Ruano FJ, Jovani R. DNA barcoding of Iberian Peninsula and North Africa Tawny Owls Strix aluco suggests the Strait of Gibraltar as an important barrier for phylogeography. Mitochondrial DNA A DNA Mapp Seq Anal. 2016; 27(6):4475-4478.
[Crossref] [Google Scholar] [PubMed]
- Fuchs J, Pons JM, Goodman SM, Bretagnolle V, Melo M, Bowie RC, et al. Tracing the colonization history of the Indian Ocean scops-owls (Strigiformes: Otus) with further insight into the spatio-temporal origin of the Malagasy avifauna. BMC Evol Biol.2008; 8(1):1-5.
[Crossref] [Google Scholar] [PubMed]
- Wang P, Yao H, Gilbert KJ, Lu Q, Hao Y, Zhang Z, et al. Glaciation-based isolation contributed to speciation in a Palearctic alpine biodiversity hotspot: evidence from endemic species. Mol Phylogenet Evol. 2018;129:315-324.
[Crossref] [Google Scholar] [PubMed]
- Renner SS. Available data point to a 4‐km‐high Tibetan Plateau by 40 Ma, but 100 molecular‐clock papers have linked supposed recent uplift to young node ages. J Biogeogr. 2016; 43(8):1479-1487.
- Lisiecki LE, Raymo ME. A Pliocene‐Pleistocene stack of 57 globally distributed benthic δ18O records. Paleoceanography. 2005; 20(1).
- Rull V. Speciation timing and neotropical biodiversity: the Tertiary-Quaternary debate in the light of molecular phylogenetic evidence. Mol Ecol. 2008; 17(11):2722-2729.
[Crossref] [Google Scholar] [PubMed]
- Rull V. Pleistocene speciation is not refuge speciation. J Biogeogr. 2015; 42(3):602-604.
- Rull V. Neotropical biodiversity: timing and potential drivers. Trends Ecol Evol. 2011; 26(10):508-513.
[Crossref] [Google Scholar] [PubMed]
- Song K, Gao B, Halvarsson P, Fang Y, Klaus S, Jiang YX, et al. Demographic history and divergence of sibling grouse species inferred from whole genome sequencing reveal past effects of climate change. BMC Ecol Evol. 2021;21:1-0.
[Crossref] [Google Scholar] [PubMed]
- Leonard JA, den Tex RJ, Hawkins MT, Muñoz‐Fuentes V, Thorington R, Maldonado JE, et al. Phylogeography of vertebrates on the Sunda Shelf: a multi‐species comparison. J Biogeogr. 2015;42(5):871-879.
- Svendsen JI, Alexanderson H, Astakhov VI, Demidov I, Dowdeswell JA, Funder S, et al. Late Quaternary ice sheet history of northern Eurasia. Quat Sci Rev. 2004; 23(11-13):1229-12271.
- Hewitt G. The genetic legacy of the Quaternary ice ages. Nature. 2000;405(6789):907-913.
[Crossref] [Google Scholar] [PubMed]
- Hewitt GM. Genetic consequences of climatic oscillations in the Quaternary. Philos Trans R Soc Lond B Biol Sci. 2004; 359(1442):183-195.
[Crossref] [Google Scholar] [PubMed]
- Kozma R, Lillie M, Benito BM, Svenning JC, Höglund J. Past and potential future population dynamics of three grouse species using ecological and whole genome coalescent modeling. Ecol Evol. 2018; 8(13):6671-6681.
[Crossref] [Google Scholar] [PubMed]
- Zhao S, Zheng P, Dong S, Zhan X, Wu QI, Guo X, et al. Whole-genome sequencing of giant pandas provides insights into demographic history and local adaptation. Nat Genet. 2013;45(1):67-71.
[Crossref] [Google Scholar] [PubMed]
- Hung CM, Shaner PJ, Zink RM, Liu WC, Chu TC, Huang WS, et al. Drastic population fluctuations explain the rapid extinction of the passenger pigeon. Proc Natl Acad Sci U S A. 2014; 111(29):10636-10641.
[Crossref] [Google Scholar] [PubMed]
- Mays HL, Hung CM, Shaner PJ, Denvir J, Justice M, Yang SF, et al. Genomic analysis of demographic history and ecological niche modeling in the endangered Sumatran rhinoceros Dicerorhinus sumatrensis. Curr Biol. 2018; 28(1):70-76.
[Crossref] [Google Scholar] [PubMed]
- Nadachowska-Brzyska K, Li C, Smeds L, Zhang G, Ellegren H. Temporal dynamics of avian populations during Pleistocene revealed by whole-genome sequences. Curr Biol. 2015; 25(10):1375-1380.
[Crossref] [Google Scholar] [PubMed]
- Provost K, Shue SY, Forcellati M, Smith BT. The genomic landscapes of desert birds form over multiple time scales. Mol Biol Evol. 2022; 39(10):msac200.
[Crossref] [Google Scholar] [PubMed]
- Claramunt S, Cracraft J. A new time tree reveals Earth history’s imprint on the evolution of modern birds. Sci Adv. 2015; 1(11):e1501005.
[Crossref] [Google Scholar] [PubMed]
- Wiens JJ. Speciation and ecology revisited: phylogenetic niche conservatism and the origin of species. Evolution. 2004;58(1):193-197.
[Crossref] [Google Scholar] [PubMed]
- Wang C, Zhao X, Liu Z, Lippert PC, Graham SA, Coe RS, et al. Constraints on the early uplift history of the Tibetan Plateau. Proc Natl Acad Sci U S A. 2008;105(13):4987-4992.
[Crossref] [Google Scholar] [PubMed]
- Pujolar JM, Blom MP, Reeve AH, Kennedy JD, Marki PZ, Korneliussen TS, et al. The formation of avian montane diversity across barriers and along elevational gradients. Nat Commun. 2022; 13(1):268.
[Crossref] [Google Scholar] [PubMed]
- Li J, Song G, Liu N, Chang Y, Bao X. Deep south-north genetic divergence in Godlewski’s bunting (Emberiza godlewskii) related to uplift of the Qinghai-Tibet Plateau and habitat preferences. BMC Evol Biol. 2019;19:1-3.
[Crossref] [Google Scholar] [PubMed]
Citation: Smith D, Roy R (2023) Mitochondrial Genome Analysis and Phylogeny and Divergence Time Evaluation of the Strix aluco. Biochem Anal Biochem.12:472.
Copyright: © 2023 Roy R, et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.