
Indexed In
- Open J Gate
- Genamics JournalSeek
- Academic Keys
- JournalTOCs
- The Global Impact Factor (GIF)
- China National Knowledge Infrastructure (CNKI)
- Ulrich's Periodicals Directory
- RefSeek
- Hamdard University
- EBSCO A-Z
- OCLC- WorldCat
- Publons
- Geneva Foundation for Medical Education and Research
- Euro Pub
- Google Scholar
Useful Links
Share This Page
Journal Flyer

Open Access Journals
- Agri and Aquaculture
- Biochemistry
- Bioinformatics & Systems Biology
- Business & Management
- Chemistry
- Clinical Sciences
- Engineering
- Food & Nutrition
- General Science
- Genetics & Molecular Biology
- Immunology & Microbiology
- Medical Sciences
- Neuroscience & Psychology
- Nursing & Health Care
- Pharmaceutical Sciences
Research Article - (2019) Volume 10, Issue 3
In Silico Design, Synthesis and Characterization of New Spebrutinib Analogues
Zaid M Jaber Al-Obaidi1*, Omar F Abdul-Rasheed2, Monther F Mahdi3 and Ayad MR Raauf42Department of Chemistry and Biochemistry, College of Medicine, Al-NahrainUniversity, Iraq
3Department of Pharmaceutical Chemistry, Ashur University College, Iraq
4Department of Pharmaceutical Chemistry, College of Pharmacy, Mustansiriyah University, Iraq
Received: 29-Jul-2019 Published: 20-Aug-2019, DOI: 10.35248/2153-2435.19.10.612
Abstract
Background: Recently, in silico or computer-aided drug design has emerged as a cornerstone on the harbor of modern drug discovery. One of the approaches to treat cancer is the inhibition of tyrosine kinase, which is considered as a key enzyme in the survival of the cancerous cells. Spebrutinib, as a member of the tyrosine kinase inhibitors, has few unwanted side effects due to its off-target bindings. In this work, the GOLD program was employed to predict the bindings and thus the inhibitory activity toward the tyrosine kinase.
Methodology: After the design and docking processes, the chemical synthesis of three spebrutinib analogues was achieved.
Results: The percent yields of the chemical syntheses were ranged from 81% to 89%. These analogues were characterized utilizing; FT-IR, DSC, CHN, and 1H NMR. In conclusion, these new spebrutinib analogues were successfully designed, synthesized, and characterized. However, these analogues are potential anticancer agents and biological activity against cancerous and toxicity pattern against normal cells are crucial to affirm the present findings.
Keywords
In silico; Computer-aided drug design; Gold; Spebrutinib analogues; Anticancer; Tyrosine kinase inhibitor
Abbreviations
CCDC: Cambridge Crystallographic Data Centre; GOLD: Genetic Optimization of Ligand Docking; TKI: Tyrosine Kinase Inhibitor; DSC: Differential Scanning Calorimeter; PDB: Protein Data Bank
Introduction
In silico drug design is a process performed to generate multiple ligand conformations and orientations, and the most appropriate ones are thereafter selected for further study [1-4]. This can be employed to fight breast cancer, which has the highest global incidence of malignancy among women, representing 25% of all cancers. Although, it has a higher mortality rate among women, especially in low-income countries [5]. In a published study, the incidence of breast cancer in Iraqi women was found to be even higher, as it represented 33.8% of all cancers registered in females aged ≥15 years in Iraq during 2000-2009, with a total of 23,792 confirmed cases [6]
Scientists have recognized tyrosine kinases as a potential target to suppress or even cure breast cancers [7]. Consequently, many tyrosine kinase inhibitors (TKI) have been developed and tested. This is exemplified by the TKI gefitinib and erlotinib. These agents possess a proved effectiveness in clinical breast cancer treatments [8-10]. However, the off-target serious side effects of these TKIs are a major obstacle that has been encountered [11-13].
In this work, the authors aimed to design, synthesize, and characterize new TKIs with the employment of the GOLD software version 5.6.3, supplied by the Cambridge Crystallographic Data Centre (CCDC).
Materials and Methods
Materials
The materials used in this work are tabulated in the Table 1.
| # | Material | Manufacturer | Country |
|---|---|---|---|
| 1 | Spebrutinib AVL-292 (99.48%) | BLDpharm | China |
| 2 | N4-(3-Aminophenyl)-5-fluoro-N2-(4-(2-methoxyethoxy) phenyl)pyrimidine-2,4-diamine (98%) | BLDpharm | China |
| 3 | Tetrahydrofuran A.R. (99%) | SCR | China |
| 4 | Potassium carbonate A.R. (99%) | SCR | China |
| 5 | Tetramethylacetyl chloride (99%) | Sigma-Aldrich | USA |
| 6 | Benzoyl chloride A.R. (99.5%) | CDH | India |
| 7 | Acetyl chloride (98%) | CDH | India |
| 8 | n-Hexane (95%) | GCC | UK |
| 9 | Dichloromethane HPLC-grade (99.8%) | GCC | UK |
| 10 | Sodium hydrogen carbonate, A.R. (99.5%) | HIMEDIA | India |
| 11 | Methanol absolute HPLC-grade | Biosolve Chimie SARL | France |
Table 1: Utilized materials with their manufacturers and countries of origin.
Instruments
The instruments used in this study are listed in the Table 2.
| # | Instrument | Manufacturer | Country |
|---|---|---|---|
| 1 | 4-digit balance | Sartorius Lab | Germany |
| 2 | Hotplate stirrer | LabTech | Korea |
| 3 | DSC (Differential Scanning Calorimeter) Thermal Analyzer | Shimadzu | Japan |
| 4 | 1-stage vacuum pump 5 Pa ¼ HP | Wenling Aitcool | China |
| 5 | Melting point apparatus | BioCote | UK |
| 6 | CHN Elemental Analyzer | EURO EA 3000 | Italy |
Table 2: Employed instruments with their manufacturers and countries of origin.
In silico docking processes
Protein preparation: The X-ray crystallographic structure of the proto-oncogene tyrosine-protein kinase (PDB ID 3CS9) protein was downloaded from the Protein Data Bank at a resolution of 2.21 Å. Then, the structure was opened with ChemBio3D Ultra 14.0 software, and energy minimization was performed. The energyminimized protein structure (Figure 1) was saved as a mol2 file and reopened in the GOLD program. In the GOLD program, the water molecules, ligands, and other hetero atoms were extracted from the protein molecule along with chains B, C and D. The addition Ligand preparation: Three best fitted and chemical synthesiscapable spebrutinib analogues were designed and synthesized. These analogues are tabulated in the Table 3.

Figure 1: The crystalline structure of the energy-minimized tyrosine kinaseFigure 1: The crystalline structure of the energy-minimized tyrosine kinase oncogene protein (PDB ID 3CS9).
| Code | IUPAC name | Formula | Chemical structure |
|---|---|---|---|
| AVL-292 | Spebrutinib: N-(3-((5-fluoro-2-((4-(2-methoxyethoxy)phenyl)amino)pyrimidin-4-yl)amino)phenyl)acrylamide. | C22H22FN5O3 | 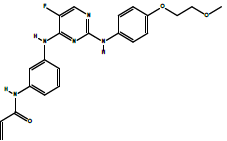 |
| 2a | N-(3-((5-fluoro-2-((4-(2-methoxyethoxy)phenyl)amino)pyrimidin-4-yl)amino) phenyl)benzamide. | C26H24FN5O3 | 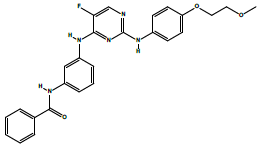 |
| 2b | N-(3-((5-fluoro-2-((4-(2-methoxyethoxy)phenyl)amino)pyrimidin-4-yl)amino) phenyl)pivalamide. | C24H28FN5O3 | 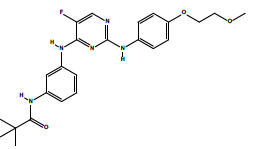 |
| 2c | N-(3-((5-fluoro-2-((4-(2-methoxyethoxy)phenyl)amino)pyrimidin-4-yl)amino)phenyl)acetamide. | C21H22FN5O3 | 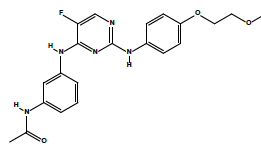 |
Table 3: The symbols, IUPAC names, chemical formulas, and the chemical structures of the spebrutinib and the synthesized analogues.
Ligand preparation: Three best fitted and chemical synthesiscapable spebrutinib analogues were designed and synthesized. These analogues are tabulated in the Table 3.
The ligand molecular structures (i.e., compounds 2a, 2b, and 2c) were drawn in ChemBioDraw Ultra version 14.0, and the energy was minimized utilizing ChemBio3D Ultra version 14.0 software. The energy-minimized ligand and protein were saved in mol2 and PDB formats, respectively, for the GOLD-assisted analysis, as explained below. All energy minimizations for the ligands were performed starting with the sketching of the two-dimensional structure and ending with the three-dimensional energy-minimized structure using ChemBioDraw Ultra 14.0 and ChemBio3D Ultra 14.0, respectively.
Docking employing GOLD software: The docking processes were performed utilizing GOLD version 5.6.3 in the present study for the prediction of the GOLD scores for the protein-ligand interactions. GOLD applies a genetic algorithm for the docking and utilizes automated docking processes to calculate the scores for the ligand and the neighborhood of the protein active site. The binding sites of the amino acid residues of the selected protein were determined by the GOLD software. This was performed for the atoms with-in 10 Å of the loaded binding residue within the binding pocket. The docked conformation that had the highest GOLD Score (fitness) was selected to analyze the mode of binding. GOLD was run, and the GOLD solutions were saved and visualized.
Chemical synthesis
The overall chemical syntheses are revealed in the Scheme 1 below.

Scheme 1: The chemical syntheses and reaction conditions of compounds 2a, 2b, and 2c.
The following chemical methods were used for the spebrutinib analogues preparations [14]:
Synthesis of compound (2-a) N-(3-((5-fluoro-2-((4-(2- methoxyethoxy)phenyl)amino) pyrimidin-4-yl) amino) phenyl) benzamide
1. Benzoyl chloride (309 mg, 2.2 mmol) was added to a stirred solution of compound (1) (665 mg, 1.8 mmol) and potassium carbonate (1.24 g, 9 mmol) in Tetrahydrofuran (THF) (12 mL) at 0°C, and the reaction mixture was stirred at 0°C for 45 min.
2. The reaction mixture was added drop-wise to a cold, solution of 10% Sodium bicarbonate (NaHCO3) (12 mL) under stirring stirred at the same temperature (0°C) for 30 min.
3. A solid precipitate was isolated by filtration and washed with cold water and hexane, and then it was dissolved in a mixture of methanol/dichloromethane (50:50, 10 mL) and concentrated under reduced pressure.
4. The residue obtained was suspended in cold water (20 mL), and Triethylamine (Et3N) was added to it, and then it was extracted with ethyl acetate (2 × 20 mL).
5. The combined ethyl acetate extract was washed with water (10 mL) and concentrated under reduced pressure in a desiccator to get 2a (0.761 mg, 89%).
Synthesis of compound (2b) N-(3-((5-fluoro-2-((4-(2-methoxyethoxy) phenyl)amino) pyrimidin-4-yl)amino) phenyl) pivalamide
1. Trimethylacetyl chloride (266 mg, 2.2 mmol) was added to a stirred solution of compound (1) (665 mg, 1.8 mmol) and potassium carbonate (1.24 g, 9 mmol) in THF (12 mL) at 0°C, and the reaction mixture was then stirred at 0°C for 45 min.
2. The reaction mixture was added drop-wise to a cold solution of 10% NaHCO3 (12 mL) under stirring and stirred at the same temperature (0°C) for 30 min.
3. A solid precipitate was isolated by filtration and washed with cold water and hexane, and then it was dissolved in a mixture of methanol/dichloromethane (50:50, 10 mL) and concentrated under reduced pressure.
4. The residue obtained was suspended in cold water (20 mL), and Triethylamine (Et3N) was added to it, and then it was extracted with ethyl acetate (2 × 20 mL).
5.The combined ethyl acetate extract was washed with water (10 mL) and concentrated under reduced pressure in a desiccator to get 2a (0.761 mg, 89%).
Synthesis of compound (2c) N-(3-((5-fluoro-2-((4-(2- methoxyethoxy)phenyl)amino)pyrimidin-4-yl)amino) phenyl)acetamide
1. Acetyl chloride (173 mg, 2.2 g, 9 mmol) was added to a stirred solution of compound (1) (665 mg, 1.8 mmol) and potassium carbonate (1.24 g, 9 mmol) in THF (12 mL) at 0°C, and the reaction mixture was stirred at 0°C for 45 min.
2.The reaction mixture was added drop-wise to a cold solution of 10% NaHCO3 (12 mL) under stirring and then stirred at the same temperature (0°C) for 30 min.
3.A solid precipitate was isolated by filtration and washed with cold water and hexane, and then it was dissolved in a mixture of methanol/dichloromethane (50:50, 10 mL) and concentrated under reduced pressure.
4.The residue obtained was suspended in cold water (20 mL), and Et3NN was added to it, and then it was extracted with ethyl acetate (2 × 20 mL).
5.The combined ethyl acetate extract was washed with water (10 mL) and concentrated under reduced pressure in a desiccator to get 2c (0.603 mg, 81%).
Results and Discussion
Results of docking
In the field of computer-aided drug design, energy minimization (aka geometry optimization, energy optimization, or geometry minimization) refers to the observation of the spatial orientation of a group of atoms in which the net force of inter-atomic origin on each atom is approximately zero [15-18]. Energy minimization is crucial to predict reliable docking results and is performed by finding a paradigm of the macromolecule and the ligand that would attach to it using a computer [19-25]. In this work, energy minimizations were performed for ligands and the target protein (i.e., the tyrosine kinase PDB code 3CS9) to be utilized further in the docking processes.
Docking of the structures: The computational docking analyses were done using GOLD) supplied by the Cambridge Crystallographic Data Centre (CCDC) (https://www.ccdc.cam. ac.uk/) as well as other related software.
The target protein is proto-oncogene tyrosine-protein kinase (PDB ID 3CS9). Spebrutinib acts as a tyrosine kinase inhibitor (TKI) along with chemicals 2a, 2b, and 2c. No constraints were used in the docking process.
The binding sites were defined as (TYR 253, VAL 256, GLU 286, and THR 315)
The In silico analyses and computational docking were successfully performed with the utilization of the GOLD program supplied by the Cambridge Crystallographic Data Centre along with other related software. The success rate for the GOLD score recorded for the GOLD program was 57% [26]. In this study, the success rate is 67%, which is considered fair for computer-aided drug design. Compound 2a shows the best fitting within the active site and the best GOLD score (better than spebrutinib) as shown in Table 4. Furthermore, compound 2a possesses hydrophobic bonding via the benzene ring within the hydrophobic pocket. Docking of the spebrutinib structure is shown in Figure 2. Docking of the 2a structure is shown in Figure 3. Docking of the 2b structure is shown in Figure 4. Docking of the 2c structure is shown in Figure 5.

Figure 2: Crystal structure of tyrosine kinase in complex with spebrutinib (PDB code: 3CS9). Spebrutinib is represented as a capped stick model, and the tyrosine kinase is shown by a spacefill representation

Figure 3: Crystal structure of the tyrosine kinase in a complex with compound 2a (PDB code: 3CS9). Compound 2a is represented as a capped stick model, and the tyrosine kinase is shown by a spacefill representation.

Figure 4:Crystal structure of the tyrosine kinase in a complex with compound 2a (PDB code: 3CS9). Compound 2a is represented as a capped stick model, and the tyrosine kinase is shown by a spacefill representation.

Figure 5 : Crystal structure of the tyrosine kinase in a complex with compound 2a (PDB code: 3CS9). Compound 2a is represented as a capped stick model, and the tyrosine kinase is shown by a spacefill representation.
| Compound Symbol | No. of Bound a.a. | No. of H-bonds | Fitness: GoldScore |
|---|---|---|---|
| Spebrutinib | 4 | 2 | 75.97 |
| 2a | 4 | 2 | 81.60 |
| 2b | 4 | 2 | 76.05 |
| 2c | 4 | 2 | 72.58 |
Table 4: shows the In silico design, the docking GOLD scores and the no. of binding sites (Amino Acid Sequence). Spebrutinib (assigned zero) is utilized as a positive control.
Results of chemical synthesis
In the DSC chart, the temperature is raised until the chemical compound reaches its melting point (Tm). The chart will spike at that temperature, as the melting process causes an endothermic change that will appear as a peak in the DSC curve. The DSC is considered as a very sensitive technique to record characteristic melting points for various analytes [27]. In this study, the melting points were recorded for spebrutinib and the synthesized compounds.
The estimated log P (Octanol-water partition coefficient) parameters, hydrogen bond donors, and hydrogen bond acceptors manifest very good compliance with the Lipinski rule of five [28].
However, the rotatable bond count is another issue that is correlated with the oral absorption feature of drugs. To have good oral bioavailability, the drug has to have no more than 10 rotatable bonds. This feature is achieved with compound 2a. In the same context, the drug likeliness is close if the topological polar surface area is equal to or less than 140 A2, which has been observed for all the synthesized compounds [29].
Regarding the tPSA, which is defined as the summation of the total polar atoms on the surface of a molecule, these atoms basically comprise oxygen and nitrogen, along with the attached hydrogens. tPSA is a widely utilized tool in medicinal chemistry to judge whether a drug is capable of permeating into cells or not. To permeate into enterocytes and become orally bioavailable, the tPSA of a molecule has to be lower than 140 square angstroms, whereas a tPSA less than 90 square angstroms renders the molecules able to penetrate the blood-brain barrier and become bioavailable to the CNS [30-32].
In chemistry, the amount of product obtained in a chemical reaction divided by the amount calculated theoretically is known as the percent yield, which is a measure of the reaction efficiency. Yields less than 40% are considered poor, more than 50% are fair, greater than 70% are considered good, greater than 80% are very good, and greater than 90% are excellent, whereas yields near 100% are known as quantitative yields [33,34]. Accordingly, the percent yields of the chemical syntheses in this work are considered very good.
Three spebrutinib analogues were successfully synthesized. Thereafter, the selected chemical and physical parameters of the synthesized compounds are tabulated in Tables 5 and 6 below:
| Compound symbol | Physical appearance | m.p. (°C)** | Log P | tPSA* |
|---|---|---|---|---|
| AVL-292 | White powder | 174.68 | 3.72 | 96.34 A2 |
| 2a | Faint yellowish-brown crystals | 148.92 | 4.94 | 96.34 A2 |
| 2b | White fluffy powder | 152.06 | 4.97 | 96.34 A2 |
| 2c | Off-white crystals | 216.76 | 3.04 | 96.34 A2 |
| *tPSA=topological polar surface area. **Melting points were observed with DSC. |
||||
Table 5: Some of the physical parameters of spebrutinib and the synthesized analogues.
| Compound symbol | Molecular mass | Yield % | H Bond Donor Count | H Bond Acceptor Count | Rotatable bond count | Heavy atom count |
|---|---|---|---|---|---|---|
| AVL-292 | 423.45 | STD | 3 | 8 | 10 | 27 |
| 2a | 473.51 | 89 | 3 | 8 | 10 | 35 |
| 2b | 453.52 | 86 | 3 | 8 | 14 | 33 |
| 2c | 411.44 | 81 | 3 | 8 | 11 | 30 |
Table 6: Selected chemical parameters of the spebrutinib standard and the synthesized compounds.
Results of characterization of the synthesized compounds
FT-IR Characterization: Structures of the synthesized compounds based on the appearance and disappearance of the characteristic bands in the observed spectra.
FT-IR Characterization of compound 2a: 3272 cm-1 (N-H stretch of amide), 1649 cm-1 (C=O stretch of amide), 1492 cm-1 (C-C stretch of aromatic), 1437 cm-1 (C-C stretch of aromatic (in-ring)).
FT-IR Characterization of compound 2b: 3432 cm-1 (N-H stretch of amide), 3364 cm-1 and 3207 cm-1 (N-H stretch of 2° amines), 1660 cm-1 (C=O stretch of amide), 1424 cm-1 (C-C stretch of aromatic (in-ring)).
FT-IR Characterization of compound 2c: 3362 cm-1 (N-H stretch of amide), 3204 cm-1 (N-H stretch of 2° amines), 1667 cm-1 (C=O stretch of amide), 1426 cm-1 (C-C stretch of aromatic (in-ring)).
Elemental Microanalysis (CHN): In general, an error no greater than 0.4% is required for these types of analyses [35]. In this study, all the synthesized compounds had errors less than 0.4% (0.21%-0.37%), which indicates a high accuracy and low content of impurities. Elemental microanalyses were performed for the spebrutinib and the synthesized compounds, and the results are tabulated in Table 7.
| Compound symbol | Chemical formula | Molecular mass | Elemental microanalyses% | ||
|---|---|---|---|---|---|
| Element | Calculated | Observed | |||
| AVL-292 | C22H22FN5O3 | 423.45 | C | 62.400 | 62.358 |
| H | 5.240 | 5.191 | |||
| N | 16.540 | 16.452 | |||
| Sum | 84.18 | 84.001 | |||
| % Deviation | 0.21% | ||||
| 2a | C26H24FN5O3 | 473.51 | C | 65.950 | 65.897 |
| H | 5.110 | 4.920 | |||
| N | 14.790 | 14.718 | |||
| Sum | 85.85 | 85.535 | |||
| % Deviation | 0.37% | ||||
| 2b | C24H28FN5O3 | 453.52 | C | 63.560 | 63.470 |
| H | 6.220 | 6.195 | |||
| N | 15.440 | 15.293 | |||
| Sum | 85.22 | 84.958 | |||
| % Deviation | 0.31% | ||||
| 2c | C21H22FN5O3 | 411.44 | C | 61.300 | 61.197 |
| H | 5.390 | 5.298 | |||
| N | 17.020 | 16.922 | |||
| Sum | 83.71 | 83.417 | |||
| % Deviation | 0.35% | ||||
Table 7: Elemental microanalyses of spebrutinib and the synthesized compounds.
1H NMR Characterization
1H NMR Characterization of compound 2a and their interpretations are shown in Table 8.
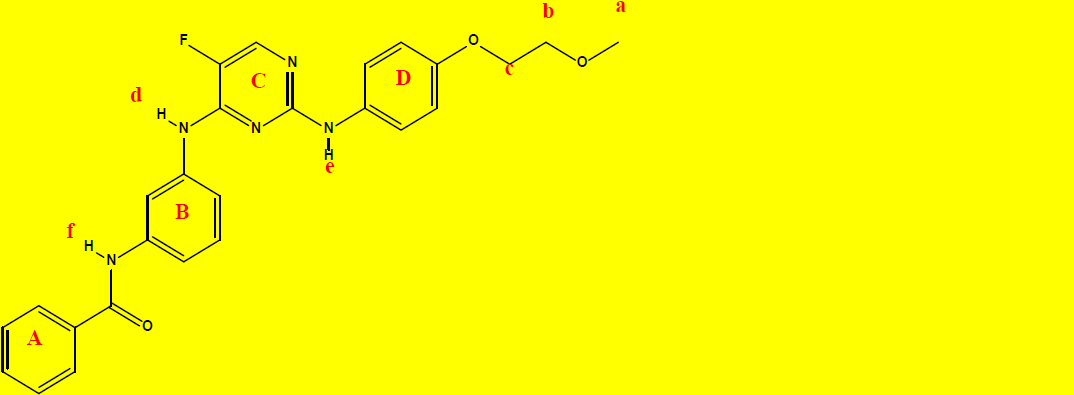 C26H24FN5O3 |
||||
| Chemical group | Chemical Shift (ppm) | Integrations | No. of Hydrogens | Interpretations |
|---|---|---|---|---|
| a | 3.32 | 3.00 | 3 | Singlet, for CH3 protons |
| b | 3.65 | 1.80 | 2 | Triplet, for CH2 protons |
| c | 4.04 | 1.94 | 2 | Triplet, for CH2 protons |
| A+B+C+D | 6.38-7.66 | 14.43 | 14 | Aromatics, rings; A, B, C, and D. |
| d | 8.06 | 1.01 | 1 | Singlet, for N-H proton as indicated. |
| e | 8.96 | 0.98 | 1 | Singlet, for N-H proton as indicated. |
| f | 9.07 | 0.98 | 1 | Singlet, for N-H proton as indicated. |
| Sum=24.14 | Total=24 | |||
Table 8: 1H NMR data and the interpretations of compound 2a.
1H NMR Characterization of compound 2b and their interpretations are shown in Table 9.
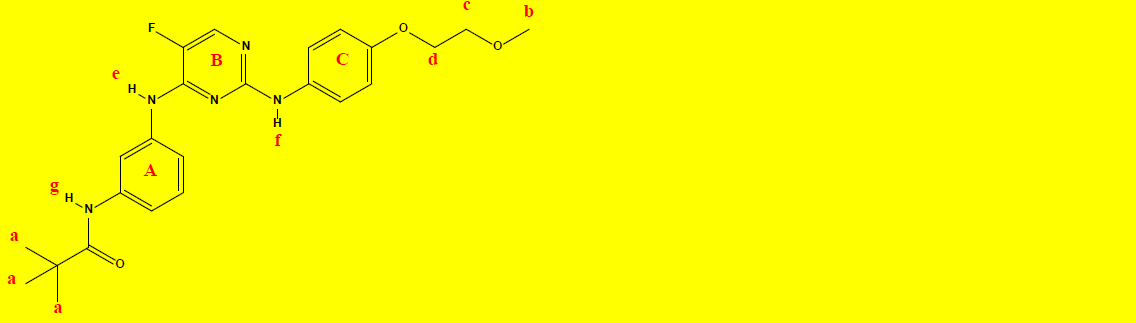 C24H28FN5O3 |
||||
| Chemical group | Chemical Shift (ppm) | Integrations | No. of Hydrogens | Interpretations |
|---|---|---|---|---|
| a | 1.16 | 8.94 | 9 | Singlet, for CH3 protons |
| b | 3.32 | 3.15 | 3 | Singlet, for CH3 protons |
| c | 3.65 | 2.13 | 2 | Triplet, for CH2 protons |
| d | 4.03 | 2.03 | 2 | Triplet, for CH2 protons |
| A+B+C | 6.63-8.09 | 9.57 | 9 | Aromatics, rings; A, B, C, and D. |
| e | 9.03 | 0.96 | 1 | Singlet, for N-H proton as indicated. |
| f | 9.21 | 0.99 | 1 | Singlet, for N-H proton as indicated. |
| g | 9.36 | 0.67 | 1 | Singlet, for N-H proton as indicated. |
| Sum=28.44 | Total=28 | |||
Table 9: 1H NMR data and the interpretations of compound 2b.
1H NMR Characterization of compound 2c and their interpretations are shown in Table 10.
 C21H22FN5O3 |
||||
| Chemical group | Chemical Shift (ppm) | Integrations | No. of Hydrogens | Interpretations |
|---|---|---|---|---|
| a | 1.78 | 2.94 | 3 | Singlet, for CH3 protons |
| b | 3.61 | 3.10 | 3 | Singlet, for CH3 protons |
| c | 3.65 | 2.03 | 2 | Triplet, for CH2 protons |
| d | 4.04 | 2.04 | 2 | Triplet, for CH2 protons |
| A+B+C | 6.79-8.09 | 9.22 | 9 | Aromatics, rings; A, B, C, and D. |
| e | 8.99 | 0.96 | 1 | Singlet, for N-H proton as indicated. |
| f | 9.37 | 0.80 | 1 | Singlet, for N-H proton as indicated. |
| g | 9.95 | 0.92 | 1 | Singlet, for N-H proton as indicated. |
| Sum=22.01 | Total=22 | |||
Table 10: 1H NMR data and the interpretations of compound 2c.
Conclusion
In this work, the authors conclude that the new spebrutinib analogues were successfully designed, synthesized, and characterized. The biological evaluation of these compounds is highly recommended.
Data Availability
“The data used to support the findings of this study are available from the corresponding author upon request”.
Conflicts of Interest
The authors confirm that there is no conflict of interest regarding the publication of this research paper.
Funding Sources
This research is self-funded.
Acknowledgment
The authors would like to acknowledge the support supplied by the College of Pharmacy, University of Kerbala, the College of Pharmacy, Mustansiriyah University, and the Iraq National Centre for Drug Control and Research for their laboratories and assistance.
REFERENCES
- Ekins S, Mestres J, Testa B. In silico pharmacology for drug discovery: Methods for virtual ligand screening and profiling. Br J Pharmacol. 2007;152:9-20.
- Bai Q, Li L, Liu S, Xiao S, Guo Y. Drug Design Progress of in Silico, in Vitro and in Vivo Researches. In-vitro In-vivo In-silico J. 2018;16:16.
- Danielson ML, Sawada GA, Raub TJ, Desai PV. In silico and in vitro assessment of OATP1B1 inhibition in drug discovery. Mol pharm. 2018;21:3060-3068.
- Khan T, Lawrence AJ, Azad I, Raza S, Joshi S, Khan AR. Computational Drug Designing and Prediction Of Important Parameters Using in silico Methods-A Review. Curr Comput Aided Drug Des. 2019.
- Alwan NA. Breast cancer among Iraqi women: Preliminary findings from a regional comparative Breast Cancer Research Project. J Glob Oncol. 2016;2:255.
- Al-Hashimi MM, Wang XJ. Breast cancer in Iraq, incidence trends from 2000-2009. Asian Pac J Cancer Prev. 2014;15:281-286.
- Burstein HJ, Elias AD, Rugo HS, Cobleigh MA, Wolff AC, Eisenberg PD, et al. Phase II study of sunitinib malate, an oral multitargeted tyrosine kinase inhibitor, in patients with metastatic breast cancer previously treated with an anthracycline and a taxane. J Clin Oncol. 2008;26:1810-1816.
- Konecny GE, Pegram MD, Venkatesan N, Finn R, Yang G, Rahmeh M, et al. Activity of the dual kinase inhibitor lapatinib (GW572016) against HER-2-overexpressing and trastuzumab-treated breast cancer cells. Cancer res. 2006;66:1630-1639.
- Moulder SL, Yakes FM, Muthuswamy SK, Bianco R, Simpson JF, Arteaga CL. Epidermal growth factor receptor (HER1) tyrosine kinase inhibitor ZD1839 (Iressa) inhibits HER2/neu (erbB2)-overexpressing breast cancer cells in vitro and in vivo. Cancer res. 2001;61:8887-8895.
- Shawver LK, Slamon D, Ullrich A. Smart drugs: Tyrosine kinase inhibitors in cancer therapy. Cancer cell. 2002;1:117-123.
- Kissick HT. Is It Possible to Develop Cancer Vaccines to Neoantigens, What Are the Major Challenges, and How Can These Be Overcome? Neoantigens as Vaccine Targets for Cancer. Cold Spring Harb Perspect Biol. 2018;10:a033704.
- Spanogiannopoulos P, Turnbaugh PJ. Broad collateral damage of drugs against the gut microbiome. Nat Rev Gastroenterol Hepatol. 2018;15:457.
- Tang LA, Dixon BN, Maples KT, Poppiti KM, Peterson TJ. Current and Investigational Agents Targeting the Phosphoinositide 3-Kinase Pathway. Pharmacotherapy. J Hum Pharmacol Drug Ther. 2018;38:1058-1067.
- https://patents.google.com/patent/ES2711249T3/enq=heteroaryl&q=compounds&q=uses&oq=heteroaryl+compounds+and+uses
- Evangelista Falcon W, Ellingson SR, Smith JC, Baudry J. Ensemble Docking in Drug Discovery: How Many Protein Configurations from Molecular Dynamics Simulations are Needed To Reproduce Known Ligand Binding. J Phys Chem B. 2019;123:5189–5195.
- Lee YV, Choi SB, Wahab HA, Lim TS, Choong YS. Applications of Ensemble Docking in Potential Inhibitor Screening for Mycobacterium tuberculosis Isocitrate Lyase Using a Local Plant Database. J Chem Inf Model. 2019;59:2487-2495.
- Martínez-Fructuoso L, Pereda-Miranda R, Rosas-Ramírez D, Fragoso-Serrano M, Cerda-Garci´a-Rojas CM, da Silva AS, et al. Structure Elucidation, Conformation, and Configuration of Cytotoxic 6-Heptyl-5, 6-dihydro-2 H-pyran-2-ones from Hyptis Species and Their Molecular Docking to a-Tubulin. J Nat Prod. 2019;82:520-531.
- Siebenmorgen T, Zacharias M. Evaluation of Predicted Protein–Protein Complexes by Binding Free Energy Simulations. J Chem Theory Comput. 2019;15:2071-2086.
- Bálint M, Horváth I, Mészáros N, Hetényi C. Towards Unraveling the Histone Code by Fragment Blind Docking. Int J Mol Sci. 2019;20:422.
- Jha P, Chaturvedi S, Swastika, Pal S, Jain N, Mishra AK. Improvising 5-HT7R homology model for design of high affinity ligands: Model validation with docking, embrace minimization, MM-GBSA, and molecular dynamic simulations. J Biomol Struct Dyn. 2018;36:2475-2494.
- Lam PC, Abagyan R, Totrov M. Ligand-biased ensemble receptor docking (LigBEnD): A hybrid ligand/receptor structure-based approach. J Comput Aided Mol Des. 2018;32:187-198.
- Padhorny D, Hall DR, Mirzaei H, Mamonov AB, Moghadasi M, Alekseenko A, et al. Protein-ligand docking using FFT based sampling: D3R case study. J Comput Aided Mol Des. 2018;32:225-230.
- Pagadala NS, Perez-Pineiro R, Bjorndahl TC, Wishart DS. Comparative molecular docking studies of EGCG with SHaPrPC. 2018;2:186–189.
- Pradeepkiran JA, Reddy PH. Structure Based Design and Molecular Docking Studies for Phosphorylated Tau Inhibitors in Alzheimer’s Disease. Cells. 2019;8:260.
- Sledz P, Caflisch A. Protein structure-based drug design: From docking to molecular dynamics. Curr Opin Struct Biol. 2018;48:93-102.
- Meng XY, Zhang HX, Mezei M, Cui M. Molecular docking: A powerful approach for structure-based drug discovery. Curr Comput Aided Drug Des. 2011;7:146-157.
- Patnaik P. Dean's analytical chemistry handbook. New York: McGraw-Hill. 2004.
- Li SK, Chantasart D. Skin Permeation Enhancement in Aqueous Solution: Correlation With Equilibrium Enhancer Concentration and Octanol/Water Partition Coefficient. J Pharm Sci. 2019;108:350-357.
- Veber DF, Johnson SR, Cheng HY, Smith BR, Ward KW, Kopple KD. Molecular properties that influence the oral bioavailability of drug candidates. J Med Chem. 2002;45:2615-2623.
- Pajouhesh H, Lenz GR. Medicinal chemical properties of successful central nervous system drugs. NeuroRx. 2005;2:541-553.
- Hitchcock SA, Pennington LD. Structure-brain exposure relationships. J med chem. 2006;49:7559-7583.
- Caron G, Ermondi G. Molecular descriptors for polarity: The need for going beyond polar surface area. Future Med Chem. 2016.
- Furniss BS. Vogel's textbook of practical organic chemistry. Pearson Education India. 1989.
- https://www.simonandschuster.com/books/MCAT-Organic-Chemistry-Review-2019-2020/Kaplan-Test-Prep/Kaplan-Test-Prep/
- Itoh N, Sato A, Yamazaki T, Numata M, Takatsu A. Determination of the carbon, hydrogen and nitrogen contents of alanine and their uncertainties using the certified reference material L-alanine (NMIJ CRM 6011-a). Anal Sci. 2013;29:1209-1212.
Citation: Al-Obaidi ZMJ, Rasheed OFA, Mahdi MF, Raauf AMR (2019) In Silico Design, Synthesis and Characterization of New Spebrutinib Analogues. Pharm Anal Acta 10:612. doi: 10.35248/2153-2435.19.10.612
Copyright: © 2019 Al-Obaidi ZMJ, et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.