
Indexed In
- Open J Gate
- Genamics JournalSeek
- Academic Keys
- JournalTOCs
- ResearchBible
- Electronic Journals Library
- RefSeek
- Hamdard University
- EBSCO A-Z
- OCLC- WorldCat
- Proquest Summons
- SWB online catalog
- Virtual Library of Biology (vifabio)
- Publons
- MIAR
- Euro Pub
- Google Scholar
Useful Links
Share This Page
Journal Flyer

Open Access Journals
- Agri and Aquaculture
- Biochemistry
- Bioinformatics & Systems Biology
- Business & Management
- Chemistry
- Clinical Sciences
- Engineering
- Food & Nutrition
- General Science
- Genetics & Molecular Biology
- Immunology & Microbiology
- Medical Sciences
- Neuroscience & Psychology
- Nursing & Health Care
- Pharmaceutical Sciences
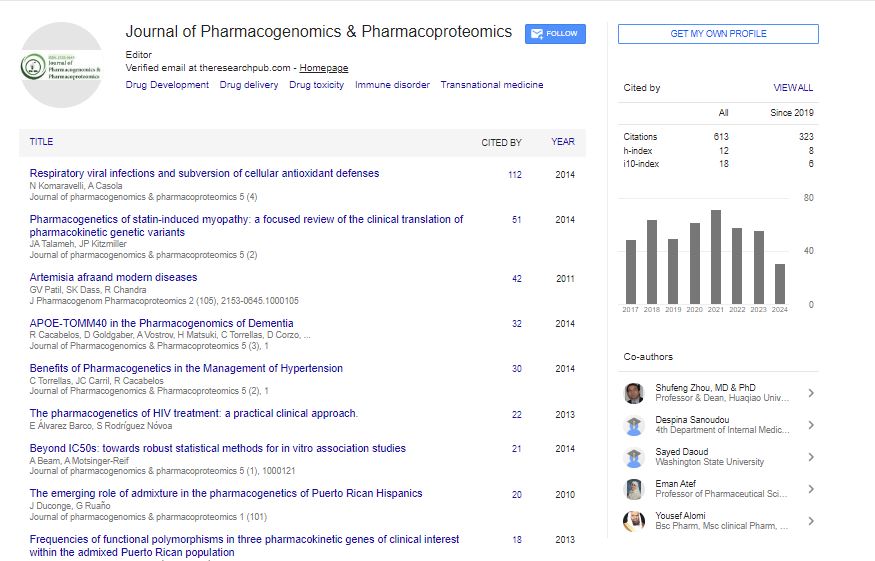
Research Article - (2023) Volume 14, Issue 3
Homemade 100 Pair Base DNA Ladder Based on Bacterial Plasmid pMAL-C2X
Firuzeh Badreh1, Khodakaram Jahanbin2, Ali Khorasani Zadeh2, Moosa Sharifat2, Milad Khayati2 and Mohammad Rashno2,3*2Department of Immunology, Ahvaz Jundishapur University of Medical Sciences, Ahvaz, Iran
3Department of Cellular and Molecular Research, Ahvaz Jundishapur University of Medical Sciences, Ahvaz, Iran
Received: 03-Dec-2022, Manuscript No. JPP-22-19104; Editor assigned: 05-Dec-2022, Pre QC No. JPP-22-19104 (PQ); Reviewed: 19-Dec-2022, QC No. JPP-22-19104; Revised: 23-Feb-2023, Manuscript No. JPP-22-19104 (R); Published: 01-Mar-2023, DOI: 10.35248/2153-0645.23.14.047
Abstract
Background: Most molecular biology research findings require markers such as DNA ladders to detect and quantify nucleic acid fragments length by electrophoresis in routine task.
Methods: In this study, we report a method to prepare a 100 pair base DNA ladder based on PCR technique. The bacterial plasmid pMAL-C2X was used as the template DNA in PCR. PCR products were extracted by phenol/chloroform, precipitated with ethanol and mixed proportionally.
Results: Finally, a set of 7 primers (5 revers and 2 forward) successfully designed. The amplified PCR fragments were quantified by nanodrop and the bands size were estimated by gel electrophoresis along with a co-migrating commercial 100 bp ladder in 1.5% agarose gel. The results indicated that the clear and sharp bands of 100 bp-1000 bp were successfully generated by one reaction in PCR.
Conclusion: Our homemade product is a competitive product with a commercial market and can be prepared with a simple and inexpensive method and low probability of non-specific band.
References
Blog | Auto Insurance in California Best Betting Sites in Netherlands Blog | Auto Insurance in USA Best Betting Sites in Morocco Blog | Car Insurance in Australia Find Lawyer in Tennessee Best Betting Sites in Romania Blog | Best Betting Sites in Bangladesh Blog | Best Betting Sites in Belgium Best Betting Sites in Germany Find Lawyer in Oregon Blog | Best Betting Sites in Spain Blog | Best Betting Sites in Ethiopia Blog | Best Betting Sites in France Find Lawyer in New Jersey Blog | Best Betting Sites in Sweden Find Lawyer in Missouri Blog - Find Lawyer in California Find Lawyer in Georgia Blog - Find Lawyer in LouisianaKeywords
Home-made; DNA ladder; Plasmid; PCR method; Primer
Introduction
DNA ladders and markers are common reagent in molecular biology and are frequently used to determine Molecular Weight (MW) or Base Pair (BP).
DNA ladder contains at least 2 or more DNA fragments of known size. Generally, samples containing DNA and DNA ladder are placed in an agarose gel in the neighboring lanes. The DNA is separated by electrophoresis through the gel, and then the gel is stained using a fluorescent dye. In fact, by comparing the bands of the sample and standard DNA Marker, the ladder can be used to estimate the unknown fragments length in routine laboratory work. DNA markers, as a prerequisite for molecular testing, are usually purchased from companies that make molecular biology materials, and there are few articles that describe how these markers are made.
However, these markers may be prepared by methods such as PCR or enzymatic digestion of bacteriophage DNA genomic or bacterial plasmids [1-3]. DNA markers were generally obtained by enzymatic digestion of E. coli plasmids or bacteriophage genomic DNA.
The arrangement of the obtained fragments depends on the type of enzyme used, the type of DNA, and the enzyme cleavage conditions. This process requires the replication of the virus or plasmid in the host and in the next step, its purification from the host and its cutting with a suitable enzyme, which is time consuming and costly and requires special facilities and equipment [4].
Unlike DNA markers, DNA ladders have multiple bands that increase in size at regular intervals, and the bands have the same resolution after staining.
Ladders are generally generated by multiplex PCR or single PCR and then the PCR products are purified and mixed together in equal concentrations [5-7]. Another way to produce DNA ladders is by enzymatic digestion of specially engineered plasmids that have one or more enzymatic cleavage sites at regular intervals [8]. In DNA ladder production using PCR, DNA pattern can be used from a variety of sources including genomic DNA of human, bacteria and viruses such as bactrophages or bacterial plasmids. But it is very important that the DNA used is easily and cheaply obtained in order to reduce the final cost of the DNA ladder and also to access it quickly and easily. Here, we describe a method to produce 100 bp DNA ladder, which include the benefits mentioned above. Based on our protocol, any laboratory can make its own 100 bp DNA ladder instead of purchasing from commercial sources.
Materials and Methods
Design of primers
To amplify optimal lengths of DNA fragment, a set of 20 oligonucleotide including 2 forward and 5 reverse primers were designed using primer 3 software. Thermodynamic details of primer dimers were considered by the primer 3 software. The corresponding expected amplified products have been showed in Table 1.
| PCR product length (bp) | Primer pairs |
|---|---|
| 100 | F1 and R1 |
| 200 | F1 and R2 |
| 300 | F1 and R3 |
| 400 | F1 and R4 |
| 500 | F1 and R5 |
| 600 | F2 and R1 |
| 700 | F2 and R2 |
| 800 | F2 and R3 |
| 900 | F2 and R4 |
| 1000 | F2 and R5 |
Table 1: Length of expected fragments based on primer pair used in this study.
Plasmid and bacterial Strain
Escherichia coli DH5 α cells and plasmid pMAL-C2X: Addgene-75286 were used as the host cell and cloning vector (the source of DNA), respectively. E. coli DH5 α harbouring the recombined vectors was cultured at 37°C on Luria Bertani (LB) agar plates supplemented with ampicillin (100 μg/ml; Sigma, USA) and incubated in a 37°C incubator for 16 hours and then plasmids were purified with a commercial Qiagen kit (QIAprep Spin Miniprep Kit-QIAGEN) according to the manufacturer instructions.
PCR reaction
PCR amplification was done in a total volume of 50 μl. Each PCR reaction consisted of 2.5 units of Taq DNA polymerase enzyme, 1 μl of dNTPs mixture (10 mM), 50 μl of each forward and reverse primer, 5 μl of the 10 × PCR buffer, 1 μl of the purified pMAL-C2X plasmid, 1.5 μl of the MgCl2 (50 mm) and 40 μl of nuclease free water PCR was performed under the following conditions: Initial denaturation at 94°C for 5 min followed by 35 cycles of at 94°C for 30 sec, annealing at 58°C for 30 sec, extension at 72°C for 30 sec, and final extension at 72°C for 7 min.
Analysis of PCR products
3 μl of PCR products were mixed with 0.5 μl of loading buffer and loaded into the 1% agarose gel. A bp 100 commercial marker was used to ensure the correct length of the amplified fragment. After loading the wells, the current of 100 volts is fixed in the electrophoresis tank. After the end of the electrophoresis, the gel was placed in the trans illuminator to observe the separated bands, and the DNA band was observed by irradiating UV light on the gel and the length of the fragment was checked (Figure 1).

Figure 1: Agarose gel electrophoresis for verification of PCR products ranging from 100 bp to 1000 bp. L: 100 bp DNA ladder, lane 1-10: 100 bp-1000 bp amplified fragments under the PCR condition without nonspecific fragments.
Ethanol precipitation of PCR products
At this stage, the PCR products were mixed with phenolchloroform- isoamyl alcohol solution (1:24:25 ratio) and vortexed for 30 seconds. After 10 minutes of centrifugation (13000 rpm), top aqueous solution containing DNA was taken and one tenth of the volume of 3 M sodium acetate solution, pH 5.2 and 2 volumes of absolute ethanol were added to it and kept overnight in the temperature was set at-20. Then a centrifugation step was performed at 4°C for 15 minutes at 13000 g, the mixture of alcohol and acetate was separated and the remaining precipitate was washed with 1 cc of 70% alcohol. Finally, the remaining sediment was dissolved using TE buffer in proportion to one tenth of the volume of the original PCR product.
Determination of PCR products concentration
The concentrate PCR products were diluted one to ten with TE buffer and 1 μl of it was determined using a nanodrop device at wavelength of 260 nm.
The purified PCR products were mixed together at certain concentrations. The final recombinant was compared with a commercial ladder (Sinaclon Co. Tehran, Iran). Then the laboratory ladder was frozen at -20°C.
Results
In the present study, a set of 7 primers (5 revers and 2 Forward) successfully were designed to generate fragments ranging from 100 bp to 1000 bp from the pMAL-C2X vector and subsequently amplicons used as a DNA ladder. In this report, appropriate primers were designed by the primer 3 software. In order to detect the successful amplification, it is important to try to avoid formation of bothering structures: Dimerization of the primers, hairpin structures, mismatching and mispriming events. Primers were produced with length of 20 bp.
Totally 10 fragments were amplified with their specific primer pairs in separated tubes that occurred under one PCR program. The amplified PCR fragments were quantified by nanodrop and the bands size were estimated by gel electrophoresis along with a co-migrating commercial 100 bp ladder in 1.5% agarose gel. The prepared DNA ladder bands were sharp and bright band pattern and could be seen under ultraviolet light. Finally, the PCR products were mixed together to make the DNA ladder via calculation of the precise concentration for every band (Figure 2).

Figure 2: A) Generation of homemade 100 bp DNA marker. All of the PCR products were combined, concentrated, and subjected to electrophoresis; B) Commercial 100 bp DNA marker (Sinaclon CO. Tehran, Iran).
Discussion
The 100 bp DNA ladders have been widely used to research and molecular laboratory. In order to limit the overall external costs, the development of homemade DNA ladder would prove to be extremely helpful. In the present study, we presented an efficient approach for the 100 bp DNA ladder production. Our method is cost effective method and simple to implement, when compared to several previously studies [9]. In the our previous finding, has been described the construction of synthetic plasmids containing restriction sites for the generation of 100 bp DNA ladder although, all fragments in vector were cloned sequentially and there was no need to purify and determine the concentration of each separate band, but cloning into vectors for transformation was a time consuming process, it needed to perform enzymatic cutting, and it also was impossible optimization of concentration of each band depending on required [10]. In order to overcome the disadvantages mentioned above, our new protocol PCR based method has been applied in fact the use of PCR is one of the relatively common methods for preparing this DNA ladder.
Although the PCR approach is simple and cost effective, the designing of the primers specific to distinct regions of the plasmid to generate 100 bp ladders is nonetheless difficult and requires several primer pairs. Actually, the need for a couple numbers of primers that are consumed during the production process and must be prepared again is disadvantage of this method.
For instance, in the study, ten primer pairs was added to a reaction system to amplify ten different fragments recently Thongnan, et al., employed PCR method for the amplification of gene fragments for production of 100 base pair DNA ladder bp using 1 universal forward primer and 11 reverse primers [11].
To resolve the limitation, to facilitate the design of primers and to optimize the experimental conditions, as well as the possibility of simultaneously performing PCR for different parts of the desired bp 100 marker, in this project, 2 forward primers and 5 reverse primers were used to produce 10 separate bands. Normally, a forward primer and a reverse primer are used to create each piece of DNA, on this basis, 10 forward primers and 10 reverse primers are needed to create 10 separate pieces, but our DNA ladder only uses 2 forward primer and 5 reverse primers have been produced, as a result, a minimum set of 7 primers designed for this study.
As expected, most of the ladder fragments were amplified easily and after optimizing the temperature of the primers was amplified in the same temperature conditions of the PCR test. Also, it is possible to change the amount of each of the bands as desired. As a result, the resolution of each of the bands can be changed to a suitable amount.
Clearly, It is a particularly useful approach for time and cost saving. In fact, the final price of the obtained product is also much lower compared to the commercial products (about one third).
Conclusion
Taken together, this study clearly introduces a rapid method and simple way to reduce costs in production of DNA ladder which is applicable for laboratory experiences. Moreover, most of the essential components for synthesizing DNA ladder were made in our own lab. Therefore, produced DNA ladder in this study is entitled as “home-made DNA ladder”.
Funding
This work was supported by a grant (CMRC-9638) from the research vice-chancellor of Ahvaz Jundishapur University of Medical Sciences, Ahvaz, Iran.
Competing Interests
No competing financial interests exist.
Author Contributions
Mohammad Rashno devised the main conceptual ideas and designed the study. Ali. khodadadi collaborated in this study as a project consultant. Material preparation was performed by Mosa. Sharifat and Milad khayati, Firuzeh baderh performed the experiments with the supervision of Mohammad Rashno. Data collection and analysis were performed by Ali. Khorasani zadeh and khoda karam. Jahanbin. The first draft of the manuscript was written by Firuzeh Badreh and all authors commented on previous versions of the manuscript. All authors read and approved the final manuscript. Mohammad rashno supervised all parts of the study and edited the article.
Ethics Approval
This is an observational study. The Ahvaz Jundishapur university of medical sciences, research ethics committee has confirmed that no ethical approval is required.
Consent to Participate
Informed consent was obtained from all individual participants included in the study.
Acknowledgement
We would like to thank the research vice-chancellor of Ahvaz Jundishapur University of medical sciences, Ahvaz, Iran for financial support and research activities throughout this study.
References
- Yasser R. Synthesis of a DNA ladder by polymerase chain reaction and optimization of yield using response surface methodology. Biotechnology. 2006;5:166-172.
- Cooney CA. Techniques and high resolution DNA size markers for pulsed field gel electrophoresis. Mol Biotechnol. 1994;2(2):119-127.
[Crossref] [Google Scholar] [PubMed]
- Polyarush SV, Egamberdiev SS, Mansurov DR, Azimova SS. Preparation of DNA markers based on E. coli plasmid DNA. Chem Nat Compd. 2003;39:592-594.
- Parker RC, Watson RM, Vinograd J. Mapping of closed circular DNAs by cleavage with restriction endonucleases and calibration by agarose gel electrophoresis. Proc Natl Acad Sci U S A. 1977;74(3):851-855.
[Crossref] [Google Scholar] [PubMed]
- Gopalakrishnan R, Joseph S, Sellappa S. Constructing a DNA ladder Range for lambdaphage by multiplex PCR. Iran J Microbiol. 2010;2(4):210.
[Google Scholar] [PubMed]
- Wang TY, Guo L, Zhang JH. Preparation of DNA ladder based on multiplex PCR technique. J Nucleic Acids. 2010;2010:421803.
[Crossref] [Google Scholar] [PubMed]
- Chang M, Wang JH, Lee HJ. Laboratory production of 100 base pair DNA molecular weight markers. J Biochem Biophys Methods. 2008;70(6):1199-1202.
[Crossref] [Google Scholar] [PubMed]
- Chen Z, Wu J, Li X, Ye C, Wenxing H. Novel strategies to construct complex synthetic vectors to produce DNA molecular weight standards. Mol Biotechnol. 2009;42:128-133.
[Crossref] [Google Scholar] [PubMed]
- Wu J, Ye C. Tandem PCR: A novel and efficient unit amplification model for the preparation of small DNA fragments. Mol Biol Rep. 2011;38:2729-2731.
[Crossref] [Google Scholar] [PubMed]
- Rashno M, Seyfi Abad Shapouri MR, Jolodar A. Construction of a synthetic vector for preparation of a 100 base pair DNA ladder. Iran J Biotechnol. 2012;10(2):106-110.
- Lertworapreecha M, Thongnan J. construction of recombinant DNA plasmid for production 100 base pair DNA ladder for endless usage based on PCR technique. Genom Genet. 2018;11(1):22-25.
Citation: Badreh F, Jahanbin K, Khodadadi A, Zadeh AK, SharifatM, Khayati M, et al. (2023) Homemade 100 Pair Base DNA Ladder Based on Bacterial Plasmid pMAL-C2X. J Pharmacogenom Pharmacoproteomics. 14:047.
Copyright: © 2023 Badreh F, et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.