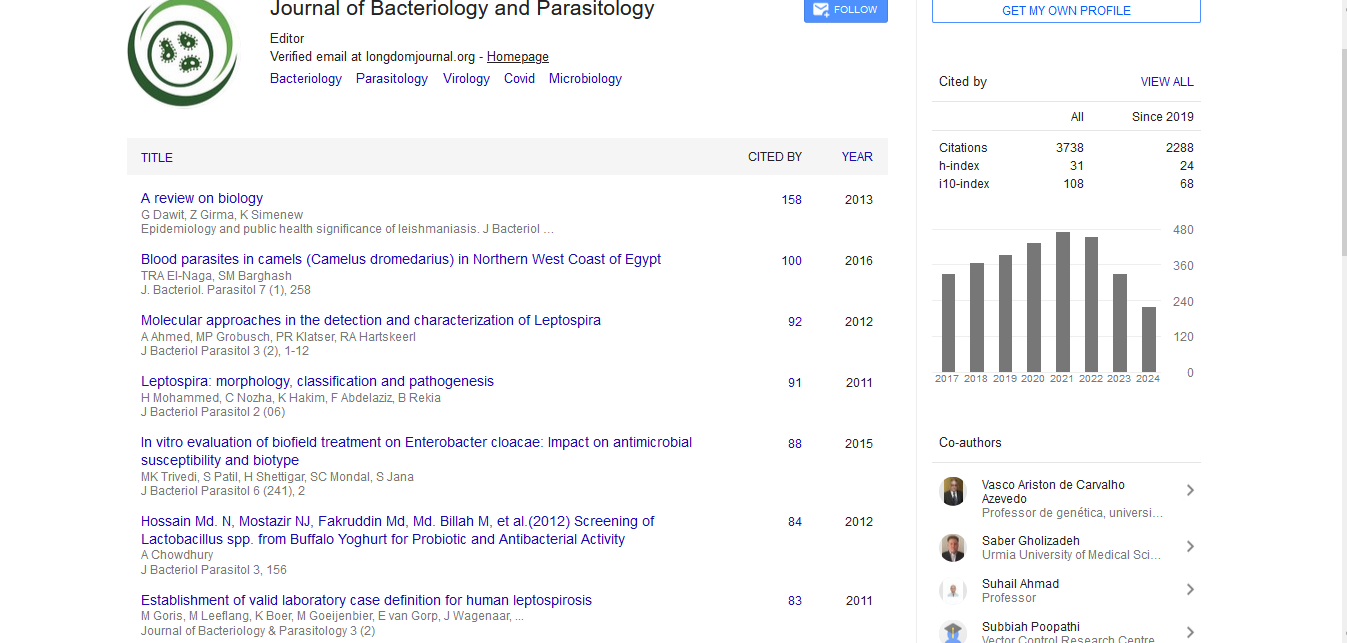

Indexed In
- Open J Gate
- Genamics JournalSeek
- Academic Keys
- JournalTOCs
- ResearchBible
- Ulrich's Periodicals Directory
- Access to Global Online Research in Agriculture (AGORA)
- Electronic Journals Library
- RefSeek
- Hamdard University
- EBSCO A-Z
- OCLC- WorldCat
- SWB online catalog
- Virtual Library of Biology (vifabio)
- Publons
- MIAR
- Geneva Foundation for Medical Education and Research
- Euro Pub
- Google Scholar
Useful Links
Share This Page
Journal Flyer

Open Access Journals
- Agri and Aquaculture
- Biochemistry
- Bioinformatics & Systems Biology
- Business & Management
- Chemistry
- Clinical Sciences
- Engineering
- Food & Nutrition
- General Science
- Genetics & Molecular Biology
- Immunology & Microbiology
- Medical Sciences
- Neuroscience & Psychology
- Nursing & Health Care
- Pharmaceutical Sciences
Research Article - (2024) Volume 15, Issue 2
Genomic Analysis of Tepidimicrobium xylanilyticum: Another Key to Lactate Metabolic Pathway in Thermophilic Anaerobic Digestion
Hou-Chia Tseng1*, Minenosuke Matsutani2, Naoshi Fujimoto1 and Akihiro Ohnishi12NODAI Genome Research Center, Tokyo University of Agriculture, Tokyo, Japan
Received: 06-Mar-2024, Manuscript No. JBP-24-25094; Editor assigned: 11-Mar-2024, Pre QC No. JBP-24-25094 (PQ); Reviewed: 25-Mar-2024, QC No. JBP-24-25094; Revised: 01-Apr-2024, Manuscript No. JBP-24-25094 (R); Published: 08-Apr-2024, DOI: 10.35248/2155-9597.24.15.505
Abstract
Tepidimicrobium xylanilyticum is a thermophilic lactate-utilising bacterium that produces H2, CO2, and acetate in pure culture using lactate as the sole carbon source. This study aimed to clarify the Thermophilic Lactate Metabolic (TLM) pathway and related genes in T. xylanilyticum participating in lactate metabolism during Thermophilic Anaerobic Digestion (TAD). De novo genome sequencing was used to determine the genomic characteristics of T. xylanilyticum. The annotations and TLM pathways of T. xylanilyticumwere predicted using DFAST and GhostKOALA. Comparative genomic analysis between the isolate EN5CB1 and type strain DSM 23310 of T. xylanilyticum was performed using CD-HIT and CLUSTALW. The genome size of T. xylanilyticum was approximately 2.9-3.1 Mbp with 33.2%-33.3% average GC-content. Through the TLM pathway, T. xylanilyticum was predicted to utilise lactate racemase, L-lactate dehydrogenases and D-lactate dehydrogenases, Pyruvate ferredoxin oxidoreductase, Pyruvate dehydrogenase complex, Phosphotransacetylase, Acetate kinase, and two types of hydrogenases. Comparative genomic analysis demonstrated higher lactate utilisation and H2 productivity in T. xylanilyticum EN5CB1 due to the insertion of a specific gene cluster and replacement of the ATPase operon. This is the first study to present the genomic characteristics and TLM pathway of T. xylanilyticum, and presents a considerable advance in our understanding of lactate metabolism in TAD.
Keywords
Anaerobic digestion; Genomic analysis, Lactate metabolic pathway; Lactate-utilising bacteria; Polylactic acid; Tepidimicrobium xylanilyticum
Introduction
The total global production of bioplastics in 2023 was approximately 2.2 million tonnes, including 52.1% biodegradable plastics, and is expected to increase to 7.4 million tons by 2028. Notably, the global production of Polylactic Acid (PLA) is expected to grow from 0.68 million tonnes (31.0%) to approximately 3.2 million tonnes (43.6%) by 2028 [1]. In our previous study, we found that PLA treatment by Thermophilic Anaerobic Digestion (TAD) was efficient and demonstrated that Lactate-Utilising Bacteria (LUB) played an important role in PLA decomposition by TAD [2]. We previously isolated Tepidimicrobium xylanilyticum EN5CB1, which could generate H2, CO2, and acetate utilising lactate as a sole carbon source in pure culture at 55°C, indicating that T. xylanilyticum is a thermophilic H2-producing LUB [3]. The microbiology of the transformation of organic matter through Anaerobic Digestion (AD) involves the cooperation of acidogenic bacteria and methanogenic archaea, which finally produce CH4 and CO2 [4]. The AD transformation pathway passes through H2, CO2, and acetate, whereas the accumulated compounds, such as lactate, may not be used directly by methanogenic archaea and must be converted by other anaerobic bacteria [4,5]. Therefore, H2- or acetate-producing LUB played a key role in AD.
Under mesophilic AD, H2-producing LUB, such as Clostridium, utilise lactate and acetate to generate butyrate, CO2, and H2 in the absence of carbohydrates [6]. The LUB Megashpaera, utilises lactate as the sole carbon source to produce H2, CO2, and acetate, and generate propionate through the acrylate pathway [7]. As an example of incomplete lactate oxidation found in Sulphate-Reducing Bacteria (SRB), Desulfovibrio vulgaris co-cultivated with methanogenic archaea, such as Methanobacterium, were reported to generate acetate and H2 from lactate in the absence of sulphate [8].
During thermophilic AD, the SRB Desulfotomaculum can degrade approximately 20 mM lactate in co-cultivation with hydrogenotrophic methanogens, whereas it cannot utilise lactate in a pure culture without sulphate [9]. Similarly, Tepidanaerobacter and Thermodesulfovibrio can degrade lactate during co-cultivation with hydrogenotrophic methanogens, such as Methanothermobacter [10,11]. In our investigations, thermophilic LUB were able to grow and utilise lactate when co-cultured with methanogens, whereas thermophilic LUB utilising lactate in pure culture have not yet been identified. Therefore, thermophilic LUB, such as T. xylanilyticum, could possibly perform lactate metabolism under thermophilic conditions in pure culture without using sulphate as electron acceptor [3,12].
Based on 16S rRNA gene sequencing, Tepidimicrobium was found to be the dominant bacteria in thermophilic anaerobic environments treated with various types of feedstock, including municipal waste sludge, food waste, manure, and AD sludge; most were suggested to be carbohydrate-metabolising bacteria, and several studies have suggested that it is the main genus related to PLA or lactate metabolism [13-17]. T. xylanilyticum type strain DSM 23310 was previously isolated, its physiological characteristics evaluated, and genome sequencing was performed (BioProject accession: PRJEB16581) [18]. However, no studies have yet investigated the lactate metabolic pathway in T. xylanilyticum during TAD.
The aim of this study was to characterise the genome and Thermophilic Lactate Metabolic (TLM) pathway of T. xylanilyticum to elucidate lactate metabolism during TAD. We also performed a comparative genomic analysis of T. xylanilyticum EN5CB1 and DSM 23310.
Materials and Methods
Bacterial strain isolation and DNA extraction
T. xylanilyticum EN5CB1 was isolated from anaerobic sludge enriched with a high concentration of lactate (5.05 g/L). The isolation methods and chromosomal DNA (Deoxyribonucleic Acid) of strain EN5CB1 were based on those previously described [3].
Sequencing library preparation, sequencing, and assembly construction
De novo genome sequencing libraries were constructed using a VAHTS Universal DNA Library Prep Kit (Vazyme Biotech, Nanjing, PRC) and loaded onto an Illumina NovaSeq 6000 Sequencing system (Illumina, San Diego, CA, USA), according to the manufacturer’s instructions, using a 2 × 150 paired-end configuration to obtain raw sequencing data (7,407,790 total reads). The quality scores (Q20 and Q30) of these sequencing data were checked based on Q Phred and trimmed using Cutadapt (v1.9.1) (Table S1) [19,20]. Sequencing data were then assembled using Velvet (version 1.2.10), and gap-filled with SSPACE (version 3.0) and GapFiller (version 1-10) for draft genome assemblies [21-23]. These Sequencing data were assembled into 39 contigs (including 3 Ns) and 36 scaffolds.
Gene predictions, annotation, and putative metabolic pathway construction
The draft genome assemblies were functionally annotated and predicted using the DDBJ Fast Annotation and Submission Tool (DFAST) prokaryotic genome annotation pipeline to obtain GC content (%), sequence coverage (depth), total number of predicted genes, Coding Sequences (CDS), and proteins [24]. The amino acid sequences predicted by DFAST were identified using NCBI BLASTP (version 2.5.0) matches against GenBank non-redundant protein sequences (nr), excluding uncultured sample sequences. Average Nucleotide Identity (ANI) analysis was performed using the OrthoANIu algorithm [25]. For metabolic pathway prediction, amino acid sequences were applied in GhostKOALA to obtain KEGG Orthology (KO) annotations and KEGG functional categories [26].
Comparative genome analysis
All protein-coding amino acid sequences from T. xylanilyticum EN5CB1 and DSM 23310 (GenBank and RefSeq) were compared and clustered using the Cluster Database at High Identity with Tolerance (CD-HIT) with a sequence overlap threshold of 70% and sequence identity of 90% [27]. Specific gene clusters detected in strain EN5CB1 were identified based on the CD-HIT pattern.
For phylogenetic analysis, amino acid sequences located in a specific gene cluster (ATPase operon) were extracted and multi-aligned using CLUSTALW (version 2.1) with default parameters [28]. The alignment results were used to construct a phylogenetic tree using the Neighbour-Joining (NJ) method in the MEGA program (version 11.0.13, Poisson model, gaps/missing data treated by pairwise deletion) with 1000 repetitions of bootstrap resampling to increase the reliability of interior branches [29,30]. Specific gene clusters were compared using GenoPlotR, and the percentage of amino acid identities was determined based on BLASTP searches [31].
Availability of data and materials
The genomic sequencing data of T. xylanilyticum EN5CB1 are registered and available at GenBank (GenBank acc. no.: BSYN01000001-BSYN01000036). For comparison, we used the genomic sequence data for T. xylanilyticum DSM 23310 (FNNG01000001-FNNG01000038), Clostridium butyricum CDC_51208 (GenBank assembly acc. no.: GCA_001886875.1), and Megasphaera elsdenii NCIMB702410 (GenBank assembly acc. no.: GCA_003006415.1), partial genome sequences of Tissierella simiarum MSJ-40 (GenBank assembly acc. no.: GCA_018917865.1), and Clostridium sp. Cult1 (GenBank assembly acc. no.: GCA_021655095.1) obtained from the NCBI Genome Database (https://www.ncbi.nlm.nih.gov/data-hub/genome/).
Results and Discussion
The genomic characteristics of T. xylanilyticum
We evaluated the draft-genome-sequence characteristics of T. xylanilyticum EN5CB1 (Table 1). The total genome sizes of strain EN5CB1 was 2,973,063 bp with average GC contents of 33.2%. We predicted 3,041 total genes, including 2,986 protein-coding genes, 2 rRNA genes, and 52 tRNA genes. We compared the draft- genome-sequence characteristics of T. xylanilyticum EN5CB1 to strain DSM 23310. Two strains showed similar total genome sizes, average GC content, and total number of genes. ANI analysis of the draft genomes showed 99.45% similarity between the strains (Figure S1). Thermophilic bacteria have genome sizes of 2-3 Mbp, suggesting that T. xylanilyticum has an average genome size [32]. Consequently, the genomic characteristics of the two strains were similar, suggesting that they are closely related.
| Genomic characteristics | EN5CB1 | DSM 23310 |
|---|---|---|
| Assembly size (bp) | 29,73,063 | 30,02,518 |
| Total number of contigs | 39 | 43 |
| GC content (%) | 33.2 | 33.3 |
| Sequence coverage (Depth) | 368X | 390X |
| Contig N50 (bp)* | 2,62,986 | 1,37,046 |
| Total number of genes | 3,041 | 3,064 |
| Protein | 2,986 | 2,969 |
| rRNA | 2 | 9 |
| tRNA | 52 | 49 |
| Other RNA | 1 | 4 |
| Sequence read archive | DRR462745 | SRR4136407 |
| GenBank accession | BSYN01000001-36 | FNNG01000001-38 |
Note: *Contig N50 (bp): length of shortest sequence among the longest sequences accounting for 50% of the entire length.
Table 1: Genomic characteristics of T. xylanilyticum EN5CB1 and DSM 23310.
Thermophilic lactate metabolic pathway of T. xylanilyticum
The predicted gene products related to lactate utilisation; H2, CO2, and acetate production; and ion transport are summarised in Table 2, and supported in Table S2. The putative TLM pathway of T. xylanilyticum was constructed as shown in Figure 1A.
| Group | Related enzyme | ||
|---|---|---|---|
| Tepidimicrobium xylanilyticum |
Clostridium butyricum |
Megasphaera elsdenii |
|
| T1 | B1 | M1 | Lactate racemase |
| T2 | B2 | M2 | Lactate dehydrogenase |
| T3 | B3 | M3 | Pyruvate-ferredoxin oxidoreductase |
| T4 | (-) | (-) | Pyruvate dehydrogenase complex |
| T5 | B4 | M4 | Phosphotransacetylase |
| T6 | B5 | M5 | Acetate kinase |
| T7 | B6 | M6 | [FeFe] Hydrogenase |
| T8 | (-) | (-) | [NiFe] Hydrogenase |
| (-) | B7 | (-) | Acetyl-CoA acetyltransferase, 3-hydroxybutyryl-CoA dehydrogenase, Crotonase, Butyryl-CoA dehydrogenase |
| (-) | B8 | (-) | Phosphate butyryltransferase, Butyrate kinase |
| (-) | B9 | (-) | Butyryl-CoA: acetate CoA transferase |
| (-) | (-) | M7 | Propionyl-CoA reductase |
| (-) | (-) | M8 | Lactyl-CoA dehydratase |
| (-) | (-) | M9 | Acrylyl-CoA reductase |
| T9 | (-) | (-) | Na (+)-translocating NADH-quinone reductase |
| T10 | (+) | (+) | Sodium proton antiporter |
| T11 | (+) | (+) | ATP synthase |
Note: T1-T11, B1-B9, and M1-M9 represent the groups of gene products related to each metabolic reaction in T. xylanilyticum, C. butyricum, and M. elsdenii, respectively. (-) and (+) represent negative and positive detections, respectively, in the genome of each microorganism.
Table 2: Enzymes or gene products of the thermophilic and mesophilic lactate metabolic pathways.

Figure 1: Predicted thermophilic lactate pathway of (A)-T. xylanilyticum and representative mesophilic lactate metabolic pathway; (B)-C. butyricum; (C)-M. elsdenii. Note: T1-T11, B1-B9, and M1-M9 represent the group of gene products related to each metabolic reaction of T. xylanilyticum, C. butyricum, and M. elsdenii, respectively. The details of each group are shown in Table 2 and Table S2.
In the TLM pathway, 53 predicted gene products were related and could be further classified into Groups T1-T11 based on their characteristics and metabolic reactions. In Group T1, L-lactate and D-lactate were potentially racemised, which was related to the lactate racemase (locus tag: EN5CB1_01870). In Group T2, conversion from L-lactate to pyruvate was enabled by L-lactate dehydrogenase (locus tag: EN5CB1_14070); similarly, conversion from D-lactate into pyruvate was related to D-lactate dehydrogenase (locus tag: EN5CB1_01710, EN5CB1_01760, EN5CB1_01860, EN5CB1_23690, and EN5CB1_23750). In Group T3, pyruvate was reversibly transformed into acetyl-CoA with the generation of CO2 by Pyruvate: Ferredoxin Oxidoreductase (PFOR; locus tag: EN5CB1_16200).
PFOR uses ferredoxin as a redox partner and transforms it into reduced Ferredoxin (Fdre), which is involved in bacterial anaerobic metabolism [33]. Similarly, in Group T4, pyruvate was transformed into acetyl-CoA and CO2; however, this reaction occurred via the utilisation of NAD+ by the Pyruvate Dehydrogenase Complex (PDHC). In EN5CB1, PDHC was encoded by two gene clusters (locus tag: EN5CB1_21680 to EN5CB1_21720 and EN5CB1_27380 to EN5CB1_27430) constructed by pyruvate dehydrogenase E1 (PDHC-E1) alpha and beta subunits, dihydrolipoamide acetyltransferase (PDHC-E2 subunit), and dihydrolipoyl dehydrogenase (PDHC-E3 subunit). In Group T5, the produced acetyl-CoA was transformed into acetyl phosphate by phosphotransacetylase (locus tag: EN5CB1_09570), and further transformed (Group T6) into acetate with the production of ATP by acetate kinase (locus tag: EN5CB1_19990 and EN5CB1_20000). [FeFe] and [NiFe] hydrogenases were detected in Group T7 and T8, respectively. In Group T7, the H2 generation process was encoded by 4Fe-4S dicluster domain containing protein (locus tag: EN5CB1_13180) and four-gene clusters including [FeFe] hydrogenase H-cluster maturation GTPase HydF (locus tag: EN5CB1_18020), [FeFe] hydrogenase H-cluster radical S-Adenosylmethionine (SAM) maturases HydG and HydE (locus tag: EN5CB1_18030 and EN5CB1_18040), and hydrogenase HydA (locus tag: EN5CB1_18050). In Group T8, we also detected the hydrogenase expression/formation protein HypE (locus tag: EN5CB1_06550), hydrogenase nickel incorporation proteins HypB and HypA (locus tag: EN5CB1_07860 and EN5CB1_07880), and hydrogenase accessory proteins HypC, HypF, and HypD (locus tag: EN5CB1_24920, EN5CB1_24930, and EN5CB1_24940). [FeFe] hydrogenase presents monomeric HydA via a set of three essential Fe-S proteins: HydE, HydF, and HydG [34]. [FeFe] hydrogenase can oxidise Fdre with H2 production in Fdox regeneration coupled with PFOR [35]. [NiFe] hydrogenase primarily involves six Hyp proteins (HypABCDEF), which have a composition similar to the [NiFe] hydrogenase in T. xylanilyticum. In addition, NAD+-reducing soluble [NiFe] hydrogenase consumes an excess of reducing equivalents by coupling the oxidation of NADH and H2 generation, resulting in NAD+ regeneration coupled with PDHC [36].
In Group T9, the sodium-translocating NADH-quinone reductase ( NA+-NQR) 6-gene cluster (locus tag: EN5CB1_12810 to EN5CB1_12860), including the ABCDEF subunit, catalyses electron transfers from NADH to quinone, coupled with the translocation of sodium ions across the membrane. In Group T10, sodium and proton antiporters (locus tag: EN5CB1_00220, EN5CB1_03710 to EN5CB1_03800), including AtpZ/AtpI family protein (AtpZ) and ATP synthase subunit I (AtpI), a (AtpB), c (AtpE), b (AtpF), delta (AtpH), alpha (AtpA), gamma (AtpG), beta (AtpD), and epsilon (AtpC), was considered to be the ATPase operon, which produces ATP from ADP and inorganic phosphate with energy supplied through the transmembrane proton motive force based on the ion gradient. The comparison between T. xylanilyticum strain EN5CB1 and DSM 23310, 49 predicted gene products were highly similar. However, it showed that 4 gene products including one lactate racemase and three D-lactate dehydrogenase was not detected in strain DSM 23310.
Based on these results, the TLM pathway of T. xylanilyticum could be characterised as follows: First, T. xylanilyticum could utilise lactate racemase to interconvert between L-lactate and D-lactate, which is then oxidised to pyruvate using L-lactate dehydrogenase or D-lactate dehydrogenase. The produced pyruvate is transformed into acetyl-CoA with CO2 generation by both PFOR and PDHC. The generated CO2 can diffuse outside the cell and acetyl-CoA can be further transformed into acetate through phosphotransacetylase and acetate kinase with ATP generation. During H2 generation, T. xylanilyticum utilises both [FeFe] and [NiFe] hydrogenase for H2 generation coupled with the regeneration of Ferredoxin ( Fdre/ Fdox) and NADH/ NAD+ to maintain the balance of the electron transport chain. Furthermore, the produced NADH along with sodium and proton ions inside and outside the cell can be balanced using transporters, such as NA+-NQR, sodium and proton antiporters, and ATP synthase. Based on previous studies, thermophilic LUB can utilise lactate in co-cultures with methanogens, whereas thermophilic lactate metabolism in pure culture has not yet been elucidated [9-11]. In the TLM pathway, L-lactate or D-lactate is converted to pyruvate with the reduction of NAD+ to NADH. Based on the previous study, reverse electron flow is thought necessary to sustain lactate oxidation to pyruvate during growth [37]. It was predicted that part of the [NiFe] hydrogenase (Group T8) consumes the excess amount of NADH, resulting in NAD+ regeneration coupled with L-lactate dehydrogenase or D-lactate dehydrogenase to keep lactate metabolism in process. This study suggests that the TLM pathway of T. xylanilyticum is a major route for lactate utilisation, and predicted H2, CO2, and acetate production in pure cultures for the first time.
We compared the TLM pathway to the representative Mesophilic Lactate Metabolic (MLM) pathway for two mesophilic LUB: C. butyricum (GCA_001886875.1) and M. elsdenii (GCA_003006415.1). The MLM pathways of C. butyricum and M. elsdenii (Figures 1B and 1C) were divided into the Groups B1-B9 and M1-M9, respectively. The MLM pathways commonly comprised lactate racemase (Groups B1 and M1), lactate dehydrogenase (Groups B2 and M2), PFOR (Groups B3 and M3), phosphotransacetylase (Groups B4 and M4), acetate kinase (Groups B5 and M5), [FeFe] hydrogenase (Groups B6 and M6), sodium proton antiporter, and ATP synthase (Table 2). Additionally, C. butyricum can produce butyrate using enzymes for acetyl-CoA transformation into butyryl-CoA (Group B7), butyryl- CoA conversion into butyrate (Group B8), and cooperation with butyryl-CoA: acetate CoA transferase (Group B9). M. elsdenii can produce propionate using propionyl-CoA reductase (Group M7), lactyl-CoA dehydratase (Group M8), and acrylyl-CoA reductase (Group M9). However, neither C. butyricum nor M. elsdenii preserved the NA+-NQR for NA+ translocation between cell membranes. These findings indicated that the TLM pathway of T. xylanilyticum and the representative MLM pathway have similar routes for lactate utilisation, along with H2, CO2, and acetate production. However, the TLM pathway of T. xylanilyticum could also utilise PDHC, [NiFe] hydrogenase for acetyl-CoA, CO2, and H2 generation, and NA+-NQR for sodium ion and NADH balancing. The findings also suggest that the TLM pathway of T. xylanilyticum is unable to transform lactate into butyrate and propionate, which varies from the MLM pathway. The AD transformation pathway passes through H2 and acetate, whereas butyrate and propionate cannot be directly utilised by methanogenic archaea [4,5]. Accordingly, the TLM pathway of T. xylanilyticum should be highly compatible with AD because of its direct generation of the main precursor for CH4 generation in methanogenic archaea.
Comparative genomic analysis of carbohydrate and lactate metabolism
According to a previous study, T. xylanilyticum EN5CB1 and DSM 23310 have different physiological characteristics, such as sugar utilisation [3]. In addition, T. xylanilyticum EN5CB1 showed approximately 3.6-fold higher lactate utilisation ability than strain DSM 23310 in pure culture, without the support of other carbon sources. We performed a comparative genomic analysis of the two strains.
The functional category classification of the protein-coding amino acid sequences of the two strains is shown in Table 3. The protein- coding amino acid sequences of EN5CB1 and DSM 23310 had similar structures, classified into 22 Groups, with approximately the same value for each category. In contrast, the number of gene products classified into carbohydrate metabolism for strain EN5CB1 was 191, which was 14% higher than that of strain DSM 23310 (167 genes). We extracted and characterised 14% (24) of the specific genes related to carbohydrate metabolism in EN5CB1 (Table S3), which were found to encode transketolase, aldolase, lactate dehydrogenase, and other carbohydrate dehydrogenases. Approximately 87.5% of these genes (21 of 24) were located in a similar location (Contig_01) in strain EN5CB1.
| KEGG functional category | EN5CB1 | DSM 23310 | |
|---|---|---|---|
| 9101 | Carbohydrate metabolism | 191 | 167 |
| 9102 | Energy metabolism | 36 | 36 |
| 9103 | Lipid metabolism | 30 | 30 |
| 9104 | Nucleotide metabolism | 66 | 66 |
| 9105 | Amino acid metabolism | 74 | 73 |
| 9106 | Metabolism of other amino acids | 22 | 22 |
| 9107 | Glycan biosynthesis and metabolism | 22 | 22 |
| 9108 | Metabolism of cofactors and vitamins | 72 | 72 |
| 9109 | Metabolism of terpenoids and polyketides | 11 | 11 |
| 9110 | Biosynthesis of other secondary metabolites | 1 | 1 |
| 9111 | Xenobiotics biodegradation and metabolism | 3 | 3 |
| 9120 | Genetic Information Processing | 157 | 169 |
| 9130 | Environmental Information Processing | 164 | 166 |
| 9140 | Cellular Processes | 82 | 82 |
| 9150 | Organismal systems | 4 | 5 |
| 9160 | Human diseases | 8 | 9 |
| 9181 | Protein families: metabolism | 49 | 50 |
| 9182 | Protein families: genetic information processing | 211 | 211 |
| 9183 | Protein families: signalling and cellular processing | 175 | 180 |
| 9191 | Unclassified: metabolism | 112 | 114 |
| 9192 | Unclassified: genetic information processing | 40 | 43 |
| 9193 | Unclassified: signalling and cellular processes | 59 | 61 |
Note: Numbers 09101-09193 represent the category number of each functional gene group defined by KEGG.
Table 3: Value and distributions of functional gene products of T. xylanilyticum.
We assessed the difference in lactate utilisation ability between the two strains. EN5CB1_1710, EN5CB1_1760, EN5CB1_1860, and EN5CB1_1870 are specific genes related to lactate utilisation and racemisation (Table S2). Therefore, we analysed the genes upstream and downstream of these four genes, from EN5CB1_01510 to EN5CB1_1910 (41 genes in total), and determined the genomic organisation (Figure 2 and Table S4).

Figure 2: Cluster-LU1 and Cluster-LU2 detected in the genome of strain EN5CB1. (A,B)-Gene sequence order in strain EN5CB1 (A, EN5CB1_01510– EN5CB1_01910) and DSM 23310 (B, SAMN05660923_01173–SAMN05660923_00431). Note: 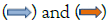 represent the homologous and specific gene sequences, respectively. The detailed annotation of EN5CB1_01510–EN5CB1_01910 is presented in Table S4.
represent the homologous and specific gene sequences, respectively. The detailed annotation of EN5CB1_01510–EN5CB1_01910 is presented in Table S4.
Except for six genes (locus tag: EN5CB1_1510, EN5CB1_1520, EN5CB1_1660, EN5CB1_1670, EN5CB1_1900, and EN5CB1_1910), the 13-gene cluster from EN5CB1_01530 to EN5CB1_01650 (Cluster-LU1) and 22-gene cluster from EN5CB1_01680 to EN5CB1_01890 (Cluster-LU2) were detected only in the genome of strain EN5CB1.
Cluster-LU1 had an average amino acid identity of 45%-77%, annotated as a Phosphotransferase System (PTS) sugar transporter, transketolase, aldolase, transcription regulator, and other unknown/ hypothetical proteins, and was closely related to Clostridium, Anaerosalibacter, Thermohalobacter, and Enterococcus. Cluster-LU2 had an average amino acid identity of 38%-86%, annotated as a PTS transporter, lactate dehydrogenase, other dehydrogenases, aldolase, lactate racemase, transketolase, and hypothetical or other proteins, and was closely related to Clostridium, Acidilutibacter, Geosporobacter, Tissierella, and Faecalicoccus. Both Cluster-LU1 and Cluster-LU2 are common associated with the PTS sugar transporter, transketolase, and aldolase, which are related to carbohydrate metabolism. However, only Cluster-LU2 presented a specific lactate dehydrogenase (locus tag: EN5CB1_01710, EN5CB1_01760, and EN5CB1_01860) and lactate racemase (EN5CB1_01870).
These clusters were closely related to different bacteria, except for Tepidimicrobium, and could possibly have originated from co- occurring bacteria by horizontal gene transfer, which is known to occur in complex environments, such as anaerobic sludge [38].
These results suggest that the insertion of Cluster-LU1 and Cluster- LU2 in a similar area may promote the expression of different carbohydrate metabolites. Furthermore, the insertion of Cluster- LU2 may explain the higher lactate utilisation ability in strain EN5CB1 compared to that in strain DSM 23310.
Comparative genomic analysis of H2 production
Compared to T. xylanilyticum DSM 23310, T. xylanilyticum EN5CB1 expressed higher H2 productivity while utilising lactate as the sole carbon source [3]. We investigated the gene products and amino acid sequences directly related to H2 generation (hydrogenase) and proton intake (ATPase operon, locus tag: EN5CB1_03710- EN5CB1_03800) by CD-HIT analysis. The two strains had an identical hydrogenase pattern, whereas a different pattern was observed for the ATPase operon (Table S5). We further analysed the ATPase operon of strain EN5CB1 (its genomic organisation in strain EN5CB1 and other closely related strains is shown in Figures 3A-3D) and found that EN5CB1_03760-EN5CB1_03800 were identical to SAMN05660923_00255-SAMN05660923_00251 of strain DSM 23310. In contrast, EN5CB1_03750 shared 77% identity with SAMN05660923_00256 and 62% identity with KQ142_ RS01835 of Tissierella simiarum. Comparing EN5CB1_03750 and SAMN02330923_00256 revealed that the upstream region had low similarity, but the downstream region was identical (Figure 3E). EN5CB1_03710 to EN5CB1_03740 showed an amino acid identity with strain DSM 23310 (locus tag: SAMN05660923_00260 to SAMN05660923_00257) of 45%-85%, whereas it was 53%-87% for T. simiarum (locus tag: KQ142_RS01855 to KQ142_RS01855). These results indicate that the upstream and downstream regions of the ATPase operon are closely related to different strains.

Figure 3: Comparison of ATPase operon detected in four strains. (A)-T. simiarumMSJ-40 (KQI42_RS01855–KQI42_RS01810); (B)-T. xylanilyticum EN5CB1 (EN5CB1_03710–EN5CB1_03800); (C)T- . xylanilyticumDSM 23310 (SAMN05660923_00260–SAMN05660923_00251); (D)-Clostridium sp. Cult1 (C3E89_RS11425–C3E89_RS11470); (E)-The amino acid sequences for EN5CB1_03750 and SAMN05660923_00256 are aligned pair-wise. Note: (*)-Represents identical amino acid sequences; (%)-Represents the amino acid identity of the four strains based on BLASTP searches.
We constructed a phylogenetic tree of amino acid sequences for this operon (Figure S2), along with one of its upstream (EN5CB1_03730) and downstream (EN5CB1_03780) regions (Figure 4), and found that EN5CB1_03730 was closely related to the F0F1 ATP synthase subunit A, located in the same clade with T. pigra, whereas T. xylanilyticum DSM 23310 was located in a completely different clade. EN5CB1_03780 was most closely related to the ATP synthase F1 subcomplex gamma subunit from T. xylanilyticum DSM 23310, whereas Tissierella occurred in a totally different clade. These results indicated that the downstream region of the ATPase operon was identical to that of strain DSM 23310; however, the upstream region was replaced with a sequence preserved in Tissierella, including T. simiarum and T. pigra. These findings strongly suggested that strains EN5CB1 and DSM 23310 have different ATPase constructs, which lead to different proton intake abilities. We hypothesised that a higher proton intake ability in strain EN5CB1 could lead to higher H2 productivity compared to that in strain DSM 23310.

Figure 4: Phylogenetic tree based on amino acid sequence alignment of (A)-EN5CB1_03730 and (B)-EN5CB1_03780. Note: Scale bar, 0.1 substitutions per amino acid site. Numerals indicate the statistical reliability of the branching order determined by bootstrap analysis (1000 duplications).
Interestingly, we also found that the ATPase operon occurred in an area similar to Cluster-LU1 and -LU2 (Contig_01). Therefore, mutations, such as insertions and replacements, occurred at similar locations in the genome of strain EN5CB1.
Strain EN5CB1 was isolated from anaerobic sludge enriched with a high lactate level (5.05 g/L), whereas strain DSM 23310 was isolated from the anaerobic sludge of a thermophilic anaerobic digester that treats municipal waste (including organic garbage, such as polysaccharide-abundant food waste) [3,39]. Therefore, strains EN5CB1 and DSM 23310 were isolated from thermophilic anaerobic sludge composed highly different carbohydrate sources. According to Galhardo et al., when bacteria are poorly adapted to their environment or stressed, the rate of mutation is promoted in their genome [40]. This suggests that, after long-term survival under TAD with high-lactate-concentration stress, strain EN5CB1 developed higher lactate utilisation and H2 production capacities owing to the insertion and replacement of specific gene clusters.
This study predicted the characteristics of the TLM pathway and related enzymes in T. xylanilyticum using genomic analyses. However, the expression of each predicted gene in the TLM pathway remain to be investigated. Additionally, the enhancement of lactate utilisation and H2 production in strain EN5CB1 requires experimental validation. We expect that RNA (Ribonucleic Acid) sequencing, gene disruption, and knockout experiments would support our findings. Nevertheless, T. xylanilyticum is a promising lactate- or PLA-decomposing enhancer in TAD.
Conclusion
This study presents the genomic characteristics of T. xylanilyticum. The TLM pathway of T. xylanilyticum and related enzymes or proteins for lactate utilisation and H2, CO2, and acetate production in pure cultures were predicted for the first time. Compared to the MLM pathway of mesophilic LUB, the TLM pathway of T. xylanilyticum is highly compatible with AD because of its direct transformation of the main precursor for CH4 generation in methanogenic archaea. Compared with strain DSM 23310, strain EN5CB1 showed higher levels of lactate utilisation and H2 production owing to the insertion and replacement of specific gene clusters and operons. Our study suggests that T. xylanilyticum is an important species for lactate metabolism in TAD.
Declaration
Acknowledgement
We would like to thank Editage (www.editage.com) for English language editing and NODAI Genome Research Center, Tokyo University of Agriculture for the genome data analysis.
Conflict of interest
The authors of this study declare no conflict of interest.
Author’s contribution
Hou-Chia Tseng: Conceptualization, investigation, writing- original draft preparation, visualization. Minenosuke Matsutani: Methodology, software, writing-review and editing. Naoshi Fujimoto: Project administration. Akihiro Ohnishi: Supervision, writing- reviewing and editing.
Data availability
The WGS of T. xylanilyticum EN5CB1 was deposited in NCBI GenBank under accession number: BSYN01000001
BioProject: PRJDB15770, BioSample: SAMD00599738
References
- European Bioplastics. Growth resumption: Global production capacities of bioplastics 2023-2028. 2023.
- Tseng HC, Fujimoto N, Ohnishi A. Biodegradability and methane fermentability of polylactic acid by thermophilic methane fermentation. Bioresour Technol Rep. 2019;8:100327.
- Tseng HC, Fujimoto N, Ohnishi A. Characteristics of Tepidimicrobium xylanilyticum as a lactate-utilising bacterium in polylactic acid decomposition during thermophilic anaerobic digestion. Bioresour Technol Rep. 2020;12:100596.
- Ziemiński K, Frąc M. Methane fermentation process as anaerobic digestion of biomass: Transformations, stages and microorganisms. Afr J Biotechnol. 2012;11(18):4127-4139.
- Moazeni F, Zhang G. Sun HJ. Imperfect asymmetry of life: Earth microbial communities prefer d-lactate but can use L-lactate also. Astrobiology. 2010;10(4):397-402.
[Crossref] [Google Scholar] [PubMed]
- Sikora A, Błaszczyk M, Jurkowski M, Zielenkiewicz U. Lactic acid bacteria in hydrogen-producing consortia: On purpose or by coincidence?. INTECH. 2013.
- Ranganathan Y, Reddy CA, Reddy A, Beveridge TJ, Breznak JA, Marzluff GA. Carbohydrate fermentations. Methods for General and Molecular Microbiology. 2007;558-585.
- Postgate JR. The sulphate-reducing bacteria. Cambridge University Press: New York. 1979.
- Klemps R, Cypionka H, Widdel F, Pfennig N. Growth with hydrogen, and further physiological characteristics of Desulfotomaculum species. Arch Microbiol. 1985;143:203-208.
- Sekiguchi Y, Imachi H, Susilorukmi A, Muramatsu M, Ohashi A, Harada H, et al. Tepidanaerobacter syntrophicus gen. nov., sp. nov., an anaerobic, moderately thermophilic, syntrophic alcohol- and lactate-degrading bacterium isolated from thermophilic digested sludges. Int J Syst Evol Microbiol. 2006;56(7):1621-1629.
[Crossref] [Google Scholar] [PubMed]
- Sekiguchi Y, Muramatsu M, Imachi H, Narihiro T, Ohashi A, Harada H, et al. Thermodesulfovibrio aggregans sp. nov. and Thermodesulfovibrio thiophilus sp. nov., anaerobic,thermophilic, sulfate-reducing bacteria isolated from thermophilic methanogenic sludge, and emended description of the genus Thermodesulfovibrio. Int J Syst Evol Microbiol. (2008;58(11):2541-2548.
[Crossref] [Google Scholar] [PubMed]
- Slobodkin A. Tepidimicrobium. In: Bergey’s Manual of Systematics of Archaea and Bacteria. Wiley. 2017;1-6.
- Lin Q, He G, Rui J, Fang X, Tao Y, Li J, et al. Microorganism-regulated mechanisms of temperature effects on the performance of anaerobic digestion. Microb Cell Fact. 2016;15:96.
[Crossref] [Google Scholar] [PubMed]
- Lin Q, De Vrieze J, Li C, Li J, Yao M, Hedenec P, et al. Temperature regulates deterministic processes and the succession of microbial interactions in anaerobic digestion process. Water Res. 2017;123:134-143.
[Crossref] [Google Scholar] [PubMed]
- Kim E, Lee J, Han G, Hwang S. Comprehensive analysis of microbial communities in full-scale mesophilic and thermophilic anaerobic digesters treating food waste-recycling wastewater. Bioresour Technol. 2018;259:442-450.
[Crossref] [Google Scholar] [PubMed]
- Cazaudehore G, Monlau F, Gassie C, Lallement A, Guyoneaud R. Methane production and active microbial communities during anaerobic digestion of three commercial biodegradable coffee capsules under mesophilic and thermophilic conditions. Sci Total Environ. 2021;784:146972.
[Crossref] [Google Scholar] [PubMed]
- Lu B, Jiang C, Chen Z, Li A, Wang W, Zhang S, et al. Fate of polylactic acid microplastics during anaerobic digestion of kitchen waste: Insights on property changes, released dissolved organic matters, and biofilm formation. Sci Total Environ. 2022;834:155108.
[Crossref] [Google Scholar] [PubMed]
- Niu L, Song L, Liu X, Dong X. Tepidimicrobium xylanilyticum sp. nov., an anaerobic xylanolytic bacterium, and emended description of the genus Tepidimicrobium. Int J Syst Evol Microbiol. 2009;59:2698-2701.
[Crossref] [Google Scholar] [PubMed]
- Ewing B, Green P. Base-calling of automated sequencer traces using phred. II. Error probabilities. Genome Res. 1998;8(3):186-194.
[Crossref] [Google Scholar] [PubMed]
- Martin M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet J 2011;17(1):10-12.
- Zerbino DR, McEwen GK, Margulies EH, Birney E. Pebble and rock band: Heuristic resolution of repeats and scaffolding in the velvet short-read de Novo assembler. PLoS One. 2009;4(12):e8407.
[Crossref] [Google Scholar] [PubMed]
- Boetzer M, Henkel CV, Jansen HJ, Butler D, Pirovano W. Scaffolding pre-assembled contigs using SSPACE. Bioinformatics. 2011;27(4):578-579.
[Crossref] [Google Scholar] [PubMed]
- Boetzer M, Pirovano W. Toward almost closed genomes with GapFiller. Genome Biol. 2012;13(6):R56.
[Crossref] [Google Scholar] [PubMed]
- Tanizawa Y, Fujisawa T, Nakamura Y. DFAST: A flexible prokaryotic genome annotation pipeline for faster genome publication. Bioinformatics. 2018;34(6):1037-1039.
[Crossref] [Google Scholar] [PubMed]
- Yoon SH, min Ha S, Lim J, Kwon S, Chun J. A large-scale evaluation of algorithms to calculate average nucleotide identity. Antonie Van Leeuwenhoek. 2017;110(10):1281-1286.
[Crossref] [Google Scholar] [PubMed]
- Kanehisa M, Sato Y, Morishima K. BlastKOALA and GhostKOALA: KEGG tools for functional characterization of genome and metagenome sequences. J Mol Biol. 2016;428(4):726-731.
[Crossref] [Google Scholar] [PubMed]
- Fu L, Niu B, Zhu Z, Wu S, Li W. CD-HIT: Accelerated for clustering the next-generation sequencing data. Bioinformatics. 2012;28(23):3150-3152.
[Crossref] [Google Scholar] [PubMed]
- Thompson JD, Higgins DG, Gibson TJ. CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 1994;22(22):4673-4680.
[Crossref] [Google Scholar] [PubMed]
- Saitou N, Nei M. The neighbor-joining method: A new method for reconstructing phylogenetic trees’. Mol Biol Evol. 1987;4(4):406-425.
[Crossref] [Google Scholar] [PubMed]
- Tamura K, Stecher G, Kumar S. MEGA11: Molecular evolutionary genetics analysis version 11. Mol Biol Evol. 2021;38(7):3022-3027.
[Crossref] [Google Scholar] [PubMed]
- Guy L, Kultima JR, Andersson SGE, Quackenbush J. GenoPlotR: Comparative gene and genome visualization in R. Bioinformatics. Oxford University Press. 2010;26(18):2334-2335.
[Crossref] [Google Scholar] [PubMed]
- Sabath N, Ferrada E, Barve A, Wagner A. Growth temperature and genome size in bacteria are negatively correlated, suggesting genomic streamlining during thermal adaptation. Genome Biol Evol. 2013;5:966-977.
[Crossref] [Google Scholar] [PubMed]
- Katsyv, Schoelmerich MC, Basen M, Müller V. The pyruvate: Ferredoxin oxidoreductase of the thermophilic acetogen, Thermoanaerobacter kivui. FEBS Open Bio. 2021;11(5):1332-1342.
[Crossref] [Google Scholar] [PubMed]
- Britt RD, Tao L, Rao G, Chen N, Wang LP. Proposed mechanism for the biosynthesis of the [FeFe] Hydrogenase H-Cluster: Central roles for the radical SAM enzymes HydG and HydE. ACS Bio and Med Chem Au. 2022;2(1):11-21.
[Crossref] [Google Scholar] [PubMed]
- Mohanraj S, Pandey A, Venkata Mohan S, Anbalagan K, Kodhaiyolii S, Pugalenthi V. Metabolic engineering and molecular biotechnology of biohydrogen production. Biohydrogen. 2019;413-434.
- Shomura Y, Taketa M, Nakashima H, Tai H, Nakagawa H, Ikeda Y, et al. Structural basis of the redox switches in the NAD+-reducing soluble [NiFe]-hydrogenase. Science. 2017;357(6354):928-932.
[Crossref] [Google Scholar] [PubMed]
- Walker CB, He Z, Yang ZK, Ringbauer JA, He Q, Zhou J, et al. The electron transfer system of syntrophically grown Desulfovibrio vulgaris. J Bacteriol. 2009;191(18):5793-5801.
[Crossref] [Google Scholar] [PubMed]
- Aminov RI. Horizontal gene exchange in environmental microbiota. Front Microbiol. 2011;2:158.
[Crossref] [Google Scholar] [PubMed]
- Wang K, Yin J, Shen D, Li N. Anaerobic digestion of food waste for volatile fatty acids (VFAs) production with different types of inoculum: Effect of pH. Bioresour Technol. 2014;161:395-401.
[Crossref] [Google Scholar] [PubMed]
- Galhardo RS, Hastings PJ, Rosenberg SM. Mutation as a stress response and the regulation of evolvability. Crit Rev Biochem Mol Biol. 2007;42(5):399-435.
[Crossref] [Google Scholar] [PubMed]
Citation: Tseng HC, Matsutani M, Fujimoto N, Ohnishi A (2024) Genomic Analysis of Tepidimicrobium xylanilyticum: Another Key to Lactate Metabolic Pathway in Thermophilic Anaerobic Digestion. J Bacteriol Parasitol. 15:505.
Copyright: © 2024 Tseng HC, et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.