


Indexed In
- Academic Journals Database
- Open J Gate
- Genamics JournalSeek
- Academic Keys
- JournalTOCs
- China National Knowledge Infrastructure (CNKI)
- CiteFactor
- Scimago
- Ulrich's Periodicals Directory
- Electronic Journals Library
- RefSeek
- Hamdard University
- EBSCO A-Z
- OCLC- WorldCat
- SWB online catalog
- Virtual Library of Biology (vifabio)
- Publons
- MIAR
- University Grants Commission
- Geneva Foundation for Medical Education and Research
- Euro Pub
- Google Scholar
Useful Links
Share This Page
Journal Flyer

Open Access Journals
- Agri and Aquaculture
- Biochemistry
- Bioinformatics & Systems Biology
- Business & Management
- Chemistry
- Clinical Sciences
- Engineering
- Food & Nutrition
- General Science
- Genetics & Molecular Biology
- Immunology & Microbiology
- Medical Sciences
- Neuroscience & Psychology
- Nursing & Health Care
- Pharmaceutical Sciences
Research Article - (2019) Volume 11, Issue 1
Biopharmaceutics Classification System (BCS) Based Biowaiver Studies of Lenalidomide Capsules (25 mg) – An Alternative to In vivo Bioequivalence Studies for Generic Oncology Drug Products
AlSwisi Mahmoud*, Boujbel Lassaad and Boujbel Mohamed AmineReceived: 08-Oct-2018 Published: 14-Feb-2019, DOI: 10.35248/0975-0851.19.11.386
Abstract
Lenalidomide, commercialized as Revlimid®, is an immunomodulatory drug, approved as a therapy for Multiple Myeloma and deletion 5q Myelodysplastic syndromes. This molecule shows a promising therapeutic potential in other hematologic malignancies. According to the European Medicines Agency (EMA), lenalidomide is considered as a fully absorbed drug substance, however, there is no sufficient data on solubility that enables its BCS classification. Therefore, as per the International Council for Harmonization (ICH) guideline, a solubility study has been done by Les Laboratoires Medis to prove that lenalidomide is a highly soluble compound. This demonstration requires the investigation in different buffer solutions within the range of pH 1- 6.8 (including pKa) at 37 ± 1°C and we have proceeded with a nominal concentration (1.0 mg/ml) which represent ten times the target concentration (0.1 mg/ml). The main reason of conducting this study is to evaluate the possibility of a BCS-based biowaiver for the consent of a generic oncology drug product unescorted by any further in vivo bioequivalence (BE) studies, also, to prove that Lenalidomide can be classified as highly soluble and highly permeable, i.e., BCS class I. The objective of establishing the BCS-based biowaiver procedure is to minimize the need for in vivo BE studies, that would reduce the exposure of healthy volunteers to oncology drugs such as lenalidomide, to prove BE. The test product and Revlimid were evaluated by in vitro methods as stated by ICH guidelines in three dissolution buffers: pH 1.2, 4.5, and 6.8, and QC release medium. The obtained results revealed that both, the test product and Revlimid have exhibited a very rapid dissolution rate (i.e. >85% in 15 min) in all dissolution buffers which infers that the test product complies with the similarity requirements for comparative dissolution testing versus the reference product. It is concluded that lenalidomide 25 mg capsules test product is bioequivalent to reference product using BCS biowaiver.
Keywords
Lenalidomide; BCS-based biowaiver; Oncology; Bioequivalence; Pharmacokinetics
Introduction
The required time for obtaining the marketing authorization depends on the development process and clinical trials. Establishing bioequivalence (BE) and bioavailability can cost up to 300.000$, not to mention the long duration to achieve, contrarily to in-vitro tests which are relatively economical (2.000$) and rapid [1]. To evaluate bioequivalence among two pharmaceutical products, the active substance in the generic product must display a similar oral bioavailability as the reference product [2]. Granting a biowaiver has become possible for immediate release (IR) solid oral drugs by means of in-vitro tests based on the Biopharmaceutics Classification System (BCS) [2]. Amidon has introduced the BCS to classify drug substances upon 3 principal factors, namely, the permeability, the aqueous solubility of the Active Pharmaceutical Ingredient (API), and the dissolution profile of the drug product [2,3]. The BCS is adopted by the US FDA as a regulatory tool to grant biowaiver, which can reduce the exposure of healthy volunteers to drugs under development. In the case of oncology drugs, the BCS-based approach grants numerous advantages when it comes to the intricacies associated with the in-vivo BE trials, especially for cytotoxic oncological drug products. It is often considered as unethical to carry out BE studies in healthy subjects using cytotoxic drugs. Therefore, the BCS-based biowaiver presents an alternative to avert the BE assessment in humans [2]. Since BCSbased biowaiver is only granted for highly soluble drug substances, the main objective of this study is to justify the merit of lenalidomide as a generic oncology drug product to be a candidate for such registration procedure.
General Characteristics
Lenalidomide is commercialized by Celgene Corporation under the name of Revlimid®. Lenalidomide (3-(4′aminoisoindoline-1′-one)-1- piperidine-2, 6-dione; Figure 1) [4], is a thalidomide derivative, a secondgeneration immunomodulatory drug, administrated orally. It has antiinflammatory, anti-angiogenic, antineoplastic and pro-erythropoietic properties [5]. The biochemical mechanism of lenalidomide consists in binding to an E3 ubiquitin ligase complex protein, cereblon, modifying its subsequent effects [6]. This interaction was demonstrated to be related to the antitumor and the immunomodulatory properties of lenalidomide [6]. The empirical formula, the gram molecular weight and other chemical properties of lenalidomide are listed in Table 1 [7]. As shown in Figure 1, lenalidomide has an asymmetric carbon atom. The optically active forms of lenalidomide are the S- and R-enantiomers produced as a 50:50 racemic mixture [6]. Lenalidomide is commercialized for oral administration as hard gelatin capsules with seven strengths: 2.5, 5, 7.5, 10, 15, 20, and 25 mg [6].
| Molecular formula | C13H13N3O3 |
| Molecular weight | 259.3 |
| Octanol-water partition coefficient | -0.43 |
| pka | 2.31 |
Table 1: Chemical proprieties of lenalidomide.

Figure 1: The structure of Lenalidomide.
Therapeutic Indication
Lenalidomide is approved as a therapy for hematologic malignancies. It's prescribed in combination with dexamethasone for the treatment of Multiple Myeloma (MM), but as a monotherapy, it's approved for the treatment of acute myeloid leukemia (AML), transfusion-dependent anemia due to Myelodysplastic Syndrome (MDS) interrelated with deletion 5q and relapsed or refractory Mantle Cell Lymphoma (MCL) [6].
Chemical Properties
Solubility study
Applying for biowaiver is restricted to drug substances performing high solubility and high or low intestinal permeability, i.e., BCS class I and BCS class III, respectively [8]. According to the European Medicines Agency (EMA) guidance, “Lenalidomide is a compound with complete absorption but the available data on solubility does not allow its BCS classification.” [9]. So as to evaluate the API’s aqueous solubility, a study ought to be led to establish the pH-solubility profile of the latter. In accordance with the ICH guideline, an active substance is categorized as high solubility if the highest single therapeutic dose is completely solubilized in 250 ml of the aqueous media at a pH range of 1.2 – 6.8 at 37 ± 1°C [8]. This demonstration requires leastways the investigation in three buffers in this range (preferably at pH 1.2, 4.5 and 6.8) and in addition at the pKa [8]. The equilibrium method was used to demonstrate the API’s solubility, it’s defined by the determination of the dose number, a dimensionless number, calculated as the equation below:
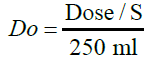
Where Do is the dose number, Dose is the highest prescribed dose in mg, S is the solubility of the API (mg/ml) and 250 ml represents the standard volume taken when an oral dosage form is administrated accordingly to the FDA guidance [10]. A dose number equal to or lower than 1 signifies high solubility, and a dose number Do >1 indicates a low- solubility compound [11].
Procedure
We conducted the study by investigating in pH: 1.2, 2.3 (pKa), 4.5, and 6.8 with a theoretical concentration of 1.0 mg/ml (100 mg in 100 ml of buffer medium) which represent ten times of the target concentration: 0.1 mg/ml (25 mg in 250 ml, 25 mg is the highest strength of Revlimid). The preparation of the buffers was realized in accordance with the European Pharmacopeia recommendations (chapter 5.17.1: Recommendations on dissolution testing). The samples were stirred during five hours using the shake flask method at 37°C.
Dissolution Study
To apply for a biowaiver, a comparative in-vitro dissolution study ought to be led [8]. In addition to HCL 0.01N dissolution medium, three Pharmacopoeia buffers should be employed: pH 1.2, 4.5, and 6.8. For BCS class I drug substances, both, the originator and the test product should exhibit either rapid or very rapid dissolution rate ≥ 85% in ≤ 30 min or ≥ 85% in ≤ 15 min, respectively, also, similar dissolution profiles under all the predetermined conditions. In case that both products exhibit different dissolution rates, the statistical similarity must be established. If both products reveal that ≥ 85% of the label amount of the drug substance is completely dissolved in 15 min, statistical evaluation is considered superfluous and the dissolution profiles are viewed as similar [8].
Pharmacokinetics Proprieties
Permeability and absorption
Permeability assessment is preferentially based on the absorption extent obtained from in vivo pharmacokinetic studies, for example, mass balance or absolute bioavailability [8]. High permeability may be appraised when absolute bioavailability is higher or equal to 85% [8]. It can be assessed as well if ≥ 85% of the administered dose is recouped unchanged in urine [8]. In healthy adults and under fasting conditions, the maximum concentration in plasma (Cmax) is observed at 0.77-1.0 h post dose, yet, in patients with AML, the estimated time to attain Cmax is 1.19 h with a standard deviation of 0.55 h after oral administration of 25 mg [4,12,13]. Higher than 90% of the dose is assessed to be absorbed in whole blood [4]. The primary elimination route is the urinary excretion where 90% of the dose was recouped unaltered in urine. Insignificant amounts of the drug were excreted in feces (4%) and in semen (0.0062%) [4]. The area under the curve (AUC) is not affected when the drug is co-administrated with food, but it reduces the Cmax [14]. In spite of that, the trials carried out for the pivotal MM registration and for the establishment of the safety and the efficiency of lenalidomide, the drug was administered without taking into consideration the food intake [15].
Distribution
Lenalidomide ex-vivo binding to plasma proteins is estimated to be low, for about 40% in healthy adults [16]. After administration of a [14C]- lenalidomide dose, the rate of radioactivity of the latter is estimated to distribute into erythrocytes for about 36-44% which is around the mean hematocrit value estimated to be 42-43%, subsequently, lenalidomide is distributed equally between plasma and cellular component of the whole blood [4], it represents the primary compound in circulation. Hence the plasma represents a suitable vehicle for the distribution of the drug in the whole blood. The range of the apparent distribution volume of [14C]-lenalidomide is often between 74-91 L in healthy volunteers, which is higher than the total volume of the water in the body (42 L) [12]. Considering the high bioavailability of lenalidomide (>90%), it is possible to surmise the existence of a restrained extravascular distribution of the drug substance. As lenalidomide is categorized as a teratogenic drug, the potential of embryo-fetal exposure, transmitted from treated males' semen into females of childbearing, must be taken into consideration [6].
Biotransformation and metabolism
Lenalidomide is subjected to a fast chiral inversion. The preferred form of lenalidomide in human is the S-form [12]. At 1 h post dose of a racemic portion, the S/R concentration ratio in plasma is settled approximately 55 and 45% of the administered dose [12]. After in-vitro studies, it is shown that lenalidomide is neither a substrate of human cytochrome P450 (CYP) enzymes nor a subject to direct conjugative metabolism [17]. Lenalidomide is degraded by a non-enzymatic hydrolysis by dint of the hydrolytic cleavage of the glutarimid ring in hepatocytes, in aqueous solutions, and in human plasma showing an in-vitro half-life of roughly 8 h [17].
Excretion and elimination
For orally administered capsules, lenalidomide is primarily eliminated as unchanged parent drug via renal excretion [6] which is deemed as rapid, due to the short plasma half-life, approximately 3 h. Subsequently, it can be inferred that even after repeated doses of lenalidomide, the plasma exhibition does not build up. For healthy adults, 300 ml/min is the total apparent clearance of the drug, 80% of it represents renal clearance and the rest (20%) is accounted for by nonrenal clearance [18]. Lenalidomide clearance is apt to decrease in older healthy volunteers, in spite of that, the drug renal excretion of it is not influenced [16].
Dosage form Performance
Excipients
Excipient variations between the reference and the test product must be evaluated for their potential to influence in-vivo absorption. Excipient quantitative and qualitative differences are allowed for BCS class I drugs, in exception of excipients that may influence the drug absorption. In this case, excipients must be quantitatively similar (± 10% of the present amount in the reference product) and qualitatively identical [8]. The used excipients in the reference product are listed in Table 2. Excipients used in the proposed test product are the same presented in the table above (Table 2). No excipients that may affect absorption (e.g., sugar-alcohols, surfactants) were used.
| Excipient | Excipient class |
|---|---|
| Anhydrous Lactose | Filler |
| Microcrystalline Cellulose | Filler |
| Croscarmelose Sodium | Disintegrant |
| Magnesium Stearate | Lubricant |
Table 2: Excipients present in Revlimid 25 mg capsules.
Results
Solubility study
The analysis of the samples was done by HPLC, using the QC assay method of the API in the drug product. The solubility results are gathered in the following Table 3.
| pH | 1,2 | 2,3 (pKa) | 4,5 | 6,8 |
| Maximum Concentration dissolved (mg/ml) | 1,01 | 0,59 | 0,43 | 0,34 |
| Final pH of samples | 1,26 | 2,29 | 3,3 | 6,79 |
| Do | 0.09 | 0.16 | 0.23 | 0.29 |
Table 3: Results of the solubility study.
Dissolution study
Figures 2-5 and Table 4 represent the comparative dissolution profiles data of the test product and Revlimid in four dissolution media, pH 1.2, 4.5 and 6.8 as per the ICH guidelines and HCL 0.01N in accordance with the US FDA dissolution method.
| Medium | Time (min) |
Lenalidomide % released | Revlimid % released |
|---|---|---|---|
| HCL 0.01N | 0 | 0 | 0 |
| 10 | 92 | 91 | |
| 15 | 98 | 95 | |
| 20 | 98 | 95 | |
| 30 | 99 | 96 | |
| 45 | 99 | 96 | |
| pH 1.2 | 0 | 0 | 0 |
| 10 | 98 | 95 | |
| 15 | 99 | 98 | |
| 2 | 100 | 99 | |
| 3 | 100 | 99 | |
| 45 | 100 | 99 | |
| pH 4.5 | 0 | 0 | 0 |
| 10 | 91 | 98 | |
| 15 | 93 | 99 | |
| 20 | 93 | 100 | |
| 30 | 94 | 100 | |
| 45 | 94 | 100 | |
| pH 6.8 | 0 | 0 | 0 |
| 10 | 90 | 92 | |
| 15 | 91 | 94 | |
| 20 | 96 | 97 | |
| 30 | 97 | 99 | |
| 45 | 97 | 99 |
Table 4: Dissolution data of lenalidomide and Revlimid 25 mg capsules.

Figure 2: Dissolution profile in HCL 0.01N.

Figure 3: Dissolution profile in pH = 1.2.

Figure 4: Dissolution profile in pH = 4.5.

Figure 5: Dissolution profile in pH = 6.8.
Discussion
Solubility study
The maximum dose strength for lenalidomide is 25 mg. A nominal concentration of 1.0 mg/ml was used to establish the solubility study which represents ten times of the target concentration of 0.1 mg/ml. Using ten times the highest single dose in this study is meant to show that the target concentration is completely dissolved and to emphasize the high solubility of the compound in order to guarantee that the highest dose (25 mg) is totally solubilized after administration of the drug. By using the equilibrium method, the results demonstrated that the compound presents a high solubility in all buffer solutions, confirming that the drug substance is completely dissolved at the target concentration of 0.1 mg/ml. Thus, the dose: solubility ratio is lower than 250 mL and Do ≤ 1, as indicated in Table 3. The drug classification based on the solubility, shows that 68% of the top 200 US list and 67% of the World Health Organization (WHO) list, are classified as highly soluble drugs based only on the dose number even if the drug has exhibited a solubility inferior to 0.1 mg/ml [11]. Consequently, we can substantiate the high solubility of the drug substance.
Dissolution study
In the present study, the dissolution test was performed in four different media to assess bioequivalence amid the test product and Revlimid. The main objective of dissolution testing is to determine the bioavailability and the in vivo performance of a drug. Generic drugs products are duplicates of inventor drug products. They are usually advocated for use because they are typically more affordable than the originator products, ergo enhancing access to life-saving medicaments, particularly in economically developing nations [1]. In case of the present study lenalidomide, 25 mg and Revlimid 25 mg capsules were studied for their bioequivalence studies. First, the dissolution was run in HCL 0.01N (Figure 2). The reference product shows a very rapid dissolution rate, 95% in 15 minutes, whereas, the test product shows a higher dissolution rate, 98% in 15 minutes. Both test and reference products exhibit a dissolution rate higher than 85% in 15 min, statistical evaluation is unnecessary in this case and the dissolution profiles are viewed as similar. Also, in pH 1.2, 4.5 and 6.8, (Figures 3-5) all the dissolution rates are considered as very rapidly dissolving, as more than 85% of the drug substance dissolved within 15 minutes. The obtained dissolution profiles are considered similar to those of the reference product in all the predetermined dissolution conditions. To sum up, any drug product that displays a very rapid dissolution rate, generally, it would not have any bioavailability issues [1].
Pharmacokinetics proprieties
Lenalidomide was rapidly absorbed under fasting condition, with Cmax observed at approximately 1 h. This high absorption rate can be explained by the very rapid dissolution rate (i.e., >85% in 15 min) shown in the dissolution assays in different pH buffers. Concomitant administration of lenalidomide with high-fat content meal does not influence the drug exhibition, calculated as AUC, yet it affects the rate and extent of the drug absorption, as revealed by an estimated 20% decrease in the AUC and an estimated reduction of 50% in Cmax together with a simultaneous delay of 1.6 h in Tmax. This delay could be explained by the food presence in the gastrointestinal tract, this mild reduction (20%) of lenalidomide oral bioavailability due to food presence is clinically considered irrelevant [19-21]. Thus, it can be inferred that lenalidomide can be taken with or without food. The absence of food effect along with the high solubility, prove that lipids have no potential effect on the solubilization and the absorption of the drug. The drug shows a high oral bioavailability (≈90%) that endorses its high permeability. It has a low protein binding, a linear pharmacokinetics, a restricted metabolism and is primarily eliminated by renal excretion as demonstrated in this study [12].
Conclusion
The objective of this study was to assess the solubility of lenalidomide in pursuance to prove its BCS class. Based on the obtained results and literature data, lenalidomide is a highly soluble and highly permeable drug substance, thus, it can be classified as BCS class I. Also, this biowaiver study has shown that our test product complies with the similarity requirements for comparative dissolution testing versus the reference product. We conclude that for lenalidomide 25 mg capsules, granting a biowaiver is justified. The biowaiver approach is meant to decrease the cost and the required time for drug approval, as well, to reduce the need for human subjects to assess bioequivalence.
Acknowledgment
Boujbel et al. are employees of Les Laboratoires Medis, which is the sponsor of the biowaiver study for lenalidomide 25 mg capsules. AlSwisi Mahmoud is an independent pharma consultant working with les Laboratoires Medis. The authors wish to acknowledge the support of Les Laboratoires Medis Research and development Department-Oncology team.
REFERENCES
- Usman S, Alam A, Nadeem N, Khawar R, Intesar Z, et al. Biowaiver Studies of Atenolol Tablets (100mg) - An Alternative to In Vivo Bioequivalence Studies. Nat Prod Chem Res. 2014;2(1):125.
- Tampal N, Mandula H, Zhang H, Li BV, Nguyen H, Conner DP. Biopharmaceutics Classification System-Based Biowaivers for Generic Oncology Drug Products: Case Studies. AAPS PharmSciTech. 2015;16(1):5-9.
- Shah VP, Amidon GL. A Theoretical Basis for a Biopharmaceutic Drug Classification: The Correlation of In Vitro Drug Product Dissolution and In Vivo Bioavailability. The AAPS J. 2014;16(5):894-898.
- Chen N, Wen L, Lau H, Surapaneni S, Kumar G. Pharmacokinetics, metabolism, and excretion of [14C]-lenalidomide following oral administration in healthy male subjects. Cancer Chemother Pharmacol. 2012;69(3):789-797.
- Syed YY, Scott LJ. Lenalidomide: A Review of its Use in Patients with Transfusion-Dependent Anaemia due to Low- or Intermediate-1-Risk Myelodysplastic Syndrome Associated with 5q Chromosome Deletion. Drugs. 2013;73(11):1183-1196.
- Chen N, Zhou S, Palmisano M. Clinical Pharmacokinetics and Pharmacodynamics of Lenalidomide. Clinical Pharmacokinetics. 2017;56(2):139-152.
- Jawhari D, Alswisi M, Ghannam M, Al-Halman J. Pharmacokinetic Comparison and Bioequivalence of a New Generic Formulation of Lenalidomide 25 mg Capsules versus Revlimid in Healthy Volunteers under Fasting Conditions. J Bioequiv Availab. 2016;8(5):214-219.
- International Council for Harmonization of Technical Requirements for Pharmaceuticals for Human Use. ICH Harmonized Guideline Biopharmaceutics Classification System-Based Biowaivers M9. 2018
- European Medicines Agency EMEA. Lenalidomide hard gelatine capsules 2.5 mg, 5 mg, 7.5 mg, 10 mg, 15 mg, 20 mg and 25 mg product-specific bioequivalence guidance. 2016.
- Food and Drug Administration. Guidance for Industry. Waiver of in vivo bioavailability and bioequivalence studies for immediate-release solid oral dosage forms based on a biopharmaceutics classification system. Rockville: Center for Drug Evaluation and Research. 2000;pp:13.
- Dahan A, Miller JM, Amidon GL. Prediction of Solubility and Permeability Class Membership: Provisional BCS Classification of the World’s Top Oral Drugs. The AAPS J. 2009;11(4): 740-746.
- Chen N, Kasserra C, Reyes J, Liu L, Lau H. Single-dose pharmacokinetics of lenalidomide in healthy volunteers: dose proportionality, food effect, and racial sensitivity. Cancer Chemother Pharmacol. 2012;70(5):717-725.
- Davies F, Baz R. Lenalidomide mode of action: linking bench and clinical findings. Blood Rev. 2010;24: S13-S19.
- Chen Y, Borthakur G. Lenalidomide as a novel treatment of acute myeloid leukemia. Expert Opin Investig Drugs. 2013;22(3):389-397.
- Celgene Europe Limited. Revlimid® (lenalidomide). Summary of product characteristics. 2018.
- Chen N, Lau H, Kong L, Kumar G, Zeldis JB, Knight R, et al. Pharmacokinetics of lenalidomide in subjects with various degrees of renal impairment and in subjects on hemodialysis. J Clin Pharmacol. 2007;47(12):1466-1475.
- Kumar G, Lau H, Laskin O. Lenalidomide: in vitro evaluation of the metabolism and assessment of cytochrome P450 inhibition and induction. Cancer Chemother Pharmacol. 2009;63(6):1171-1175.
- Chen N, Weiss D, Reyes J, Liu L, Kasserra C, Wang X, et al. No clinically significant drug interactions between lenalidomide and P-glycoprotein substrates and inhibitors: results from controlled phase I studies in healthy volunteers. Cancer Chemother Pharmacol. 2014;73(5):659-660.
- List A, Dewald G, Bennett J, Giagounidis A, Raza A. Myelodysplastic Syndrome-003 Study Investigators. Lenalidomide in the myelodysplastic syndrome with chromosome 5q deletion. N Engl J Med. 2006;355:1456-1465.
- Dimopoulos M, Spencer A, Attal M, Prince HM, Harousseau JL. Multiple Myeloma (010) Study Investigators. Lenalidomide plus dexamethasone for relapsed or refractory multiple myeloma. N Engl J Med. 2007;357:2123-2132.
- Weber DM, Chen C, Niesvizky R, Wang M, Belch A. Multiple Myeloma (009) Study Investigators. Lenalidomide plus dexamethasone for relapsed multiple myeloma in North America. N Engl J Med. 2007;357:2133-2142.
Citation: Mahmoud A, Lassaad B, Amine BM (2019) Biopharmaceutics Classification System (BCS) Based Biowaiver Studies of Lenalidomide Capsules (25 mg) – An Alternative to In vivo Bioequivalence Studies for Generic Oncology Drug Products. J Bioequiv Availab 11:386. doi: 10.35248/0975-0851.19.11.386
Copyright: © 2019 Mahmoud A, et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.