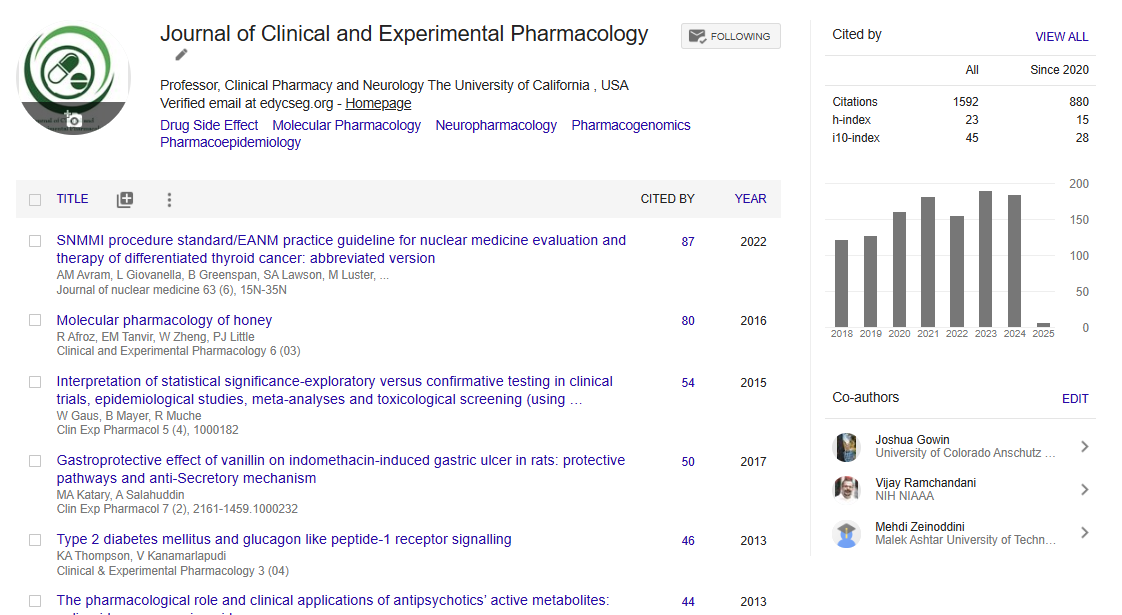

Indexed In
- Open J Gate
- Genamics JournalSeek
- China National Knowledge Infrastructure (CNKI)
- Ulrich's Periodicals Directory
- RefSeek
- Hamdard University
- EBSCO A-Z
- OCLC- WorldCat
- Publons
- Google Scholar
Useful Links
Share This Page
Journal Flyer

Open Access Journals
- Agri and Aquaculture
- Biochemistry
- Bioinformatics & Systems Biology
- Business & Management
- Chemistry
- Clinical Sciences
- Engineering
- Food & Nutrition
- General Science
- Genetics & Molecular Biology
- Immunology & Microbiology
- Medical Sciences
- Neuroscience & Psychology
- Nursing & Health Care
- Pharmaceutical Sciences
Review Article - (2023) Volume 13, Issue 6
A Systemic Review on Aducanumab and Lecanamab in the Development of ARIA in Alzheimer's Disease
Ankit Verma*, Roshi and Priyanshi SharmaReceived: 25-Sep-2023, Manuscript No. CPECR-23-23179; Editor assigned: 27-Sep-2023, Pre QC No. CPECR-23-23179(PQ); Reviewed: 11-Oct-2023, QC No. CPECR-23-23179; Revised: 18-Oct-2023, Manuscript No. CPECR-23-23179(R); Published: 25-Oct-2023, DOI: 10.35248/2161-1459.23.13.393
Abstract
Alzheimer's is a condition of neurodegenerative disease, which caused cognitive and memory loss in the patient. The condition usually progresses in those over 65, and it causes dementia to develop along with memory loss and impairments in cognitive behavior. By 2050, there will be 113 million cases of dementia worldwide, up from the 50 million cases there were in 2010. Amyloid plaques and neurofibrillary tangles are the major cause of AD. Formation of Aβ associated with the conversion of APP protein into Aβ through γ-secretase enzyme and formation of neurotangillary fibre are associated with hyperphosphorylation of tau protein. Disease modifying therapy like aducanumab, bapineuzumab, gantenerumab, donanemab, crenezumab, solanezumab and lecanamab are developed. It is believed that these Anti Aβ antibodies are very significantly reduce the level of Aβ. Aducanumab and lecanamab showing good therapeutic effect with lesser degree of side effect. ARIA-A and ARIA-H are most commonly associated side effect of these anti Aβ antibody. In this review we are focused on the comparison between Aducanumab and lecanamab, their mechanism of reducing Aβ level, binding profile with soluble and insoluble plaque and their side effect.
Keywords
Alzheimer disease; Amyloid Beta; APP gene; Aducanumab; Lecanamab; Neurofibrillary tangles; Tau protein
Introduction
Alzheimer’s disease ia s neurodegnerative disease, in which person lost his memory power and cognitive function. Progression of this disease generally seen in the person aged >65years, and it leads to development of dementia accompanied by memory loss and impairment in cognitive behavior [1]. It is a major problem occurs worldwide and it will estimate that 115 millions of people, who is >65 years or above are affected by Alzheimer’s disease [2]. Age is the major risk factor which is particularly seen in the development of AD and also in dementia [3]. Women>65 years are of greater risk than men having dementia owing to an excess all-cause mortality in men aged>45 years [4]. But in many study find that there is no any difference in both sexes in development of dementia [5].
Mutation in gene like APP, PSEN1, PSEN2 are rare genetic risk factor for AD. Person who have inherited these mutated gene are almost younger than 65 years when they express symptoms of AD (Figure 1) [6]. A new and rare variant of APP gene (A673T) is identified as a protective against Aβ production [7]. The identifiable mechanism involved in development of AD is plaque of Amyloid β(Aβ) and formation of neurofibrillary tangle inside the neuronal cell. This is developed due to hyper phosphorylation of Tau protein [8,9]. Tau protein is responsible for microtubule stabilization, present normally in cytoplasm and also in both presynaptic and post-synaptic compartment depending on the splicing of exon they are usually occurs in six isoform and they are known as 3R and 4R isoform due to three or four microtubule binding domain [3,10].

Figure 1: Schematic diagram represent risk factor associated with Alzheimer’s disease development.
Post translational modification in tau cause increase its phosphorylation capacity up to 3-4 times than normal phosphorylation [9]. This hyperphosphorylated altered conformational tau, aggregated into paired helical filament in cell body and dendrite and form neurofibrillary tangles (intracellular tau aggregates), neuropil threads (tau fragments in the neuropil) and dystrophic neurites (tau-containing degenerated axons and dendrites surrounding Aβ plaques [8]. A neurotoxic fragment of Tau protein (NH2hTau), is mapped between 26 and 230 amino acid of the longest human tau isoform, damage the mitochondria by mitophagy process [11]. In mature hippocampal neuron, expression of these neurotoxic fragment leads to synaptic alteration, mitochondrial structure alteration and enhance mitophagic flux [12]. Neuron in entorhinal cortex and hippocampus region is mainly degenerated and due to formation of neurofibrillary tangle, the message conveying pathway is blocked [13]. Amyloid Precursor Protein (APP) is degraded by β-and γ-secretase (known as amyloidogenic pathway) and form Aβ peptides like Aβ40, Aβ42 and Aβ43. These fragments of Aβ stick with each other and form insoluble β-sheet fibrillar aggregates. These aggregate are then deposited into many area in the brain like extracellular parenchyma, cerebral vasculature [14]. These deposition cause synaptic damage and alter the functions. Neuronal atrophy is developed in hippocampus area and then spreading to the cortical region that results in cognitive impairment and dementia [13]. Aβ exist in many forms like monomer, soluble aggregate of different size (oligomers and protofibrils) and insoluble fibrils [15]. Study data of soluble protofibrils indicates that it is neurotoxic in nature and contribute to the damage of neuron [16,17]. Along with the Aβ formation in amyloidogenic pathway, other molecule like APPSβ, βCTF and AICD are also formed (Figures 2 and 3) [18]. Apolipoprotein E encoded by APOE gene, produced in brain by astrocyte and microglia believed to cause increased risk of AD by affecting the onset of Aβ aggregation in the brain by altering the clearance and seeding of Aβ (Figure 4) [19,20]. The precise mechanisms by which Aβ/APP and tau interact are not well understood.

Figure 2: Diagrammatic representation demonstrate the action of α-secretase on APP and form APPsα, αCTF and γ-secretase enzyme act on α-CTF leads to formation of AICD and p3.

Figure 3: Diagrammatic representation demonstrate the action of β-secretase enzyme on APP and form APPsβ, βCTF, γ- secretase. Enzyme act on β-CTF leads to formation of AICD and Aβ.

Figure 4: Schematic representation show the formation of Aβ plaque from APP in the presence of β-secretase enzyme. In this pathways APP is converted into APPsβ and βCTF by β- Secretase and on βCTF enzyme γ-secretase act and form soluble Aβ, these soluble form converted into monomer form and then into oligomer to fibrils and finally into plaques.
Diagnosis of AD
Positron Emission Tomography (PET) technique is widely used for the detection of AD. PET detect and analyse tracer molecule (Aβ, tau protein) and CSF protein present in excessive amount in brain cell and CSF [21]. Phosphorylated Tau 181 protein and Fibrillar Aβ is considered as an important biomarker of AD [22]. These two biomarker are also present in another disease and it can be detected in-vivo or in-vitro by scanning tunneling microscopy (for Aβ) and photon rayleigh scattering assay (for tau detection) [23]. Other one of the major neuroinflammation target for PET scanning is Translocator Protein (TSPO). TSPO is mainly found in mitochondrial outer membrane and steroid synthesizing, it is responsible for transportation of cholesterol into mitochondria [21]. Up regulation of these transporter protein gives an evidence of AD. Different generation of TSPO identifier are used in detection of AD.
1st generation TSPO identifier molecules
(C11)PK-11195 is the first generation widely used identifier molecules. It gives low signal to noise ration in final PET image because of low permeability of blood brain barrier and high non-specific plasma binding [24,25].
2nd generation TSPO identifier molecules
Due to some problems in 1st generation of TSPO identifier, few 2nd generation identifier including (11C)DAA1106, (1F) FEDAA1106, (125I)CLINDE (11C)PBR06, (11C)PBR28, (18F)PBR111, (18F)DPA-713, (18F)DPA-714, (18F)F-DPA, (11C)AC5216, (18F)FEMPA, and (18F)FEPPA are used [26,27]. Due to the polymorphism TSPO (rs6971) each of the TSPO identifier has different binding affinity.
3rd generation TSPO identifier molecule
In this, (18F)GE-180, (R,S)-(18F)GE-387, (11C)ER176, (11C)CB184, (11C)CB190, (11C)N′-MPB, and (18F)LW223 these molecules are used as TSPO identifier. In this (18F)GE-180 (flutriciclamide), (S)-(18F)GE-387, and (11C) ER176 resolve the problem of ligand-dependent affinity in in-vitro binding assay where these identifier are insensitive to TSPO rs6971 polymorphisms [21,28].
Some of the other identification target like monoamine oxidase-B (MAO-B), matrix metalloproteinases, colony-stimulating factor 1 receptor (CSF1R), imidazoline-2 binding sites (I2BS), cyclooxygenases, the phospholipase A2/arachidonic acid pathway, sphingosine-1-phosphate receptor-1, reactive oxygen species, cannabinoid-2 receptor, purinergic P2X7 receptor and P2Y12 receptor, the fractalkine receptor CX3CR1 (187), TREM2 (140), and receptor for advanced glycation end products are under development [29-32].
Treatment Approach
Till now, no any approved drug is available in the market for the fully curable treatment of AD. Drug which is available, reduces the symptom of AD at desired level so patient feel some comfortable [33]. Drugs such acetylcholinestrase inhibitors (Donopezil, Galantamine, rivastigmine), N-methyl-D-aspartate receptor antagonist (Memantine) are used for sympatomatic relief of AD patient [34,35]. Based on the Aβ mechanism, it is believed that clearance of Aβ plaques may be useful approach in reduction of progression of AD [36-38]. Anti-Aβ drug are made for this purpose and scientist till now try to find out exact molecule which can reduce the AD progression but unfortunately, all effort made to find out a drug molecule has been failed in the past [39]. Growing evidences in the field of immune system, innate immune plays an important role in disease progression and AD etiology, CNS has considered as the immune-privileged site [40,41]. Immunotherapy is an emerging approach for the treatment of AD. Some of the Anti-Aβ antibodies like aducanumab,bapineuzumab, gantenerumab, donanemab crenezumab, solanezumab and lecanamab are developed and already goes into clinical trials [42]. These Antibodies target the Aβ through vatious metabolic pathway to remove it. Bapineuzumab has high affinity for all form of Aβ ,but it has been terminated from clinical trial because it does not produce desired effects [43]. Solanezumab has mid-range soluble monomeric Aβ anti Aβ antibodies, it has been terminated from clinical trials in phase 3 because it doesn’t meet essential criteria endpoint [44].
Sympatomatic relieving drugs
Acetylcholine relieving drugs. E.g.:-Donepezil, galantamine, rivastigmine
N-methyl-D-ASPARATE receptor antagonist. E.g.:- Memantine
Anti-Aβ Antibodies
Aducanumab, lecanamab, gantenerumab, donanemab, crenezumab
Aducanumab: Aducanumab is categorized under IgG 1, which is a human immunoglobulin. It significantly reduces the Aβ aggregate by selectively targeting the aggregated Aβ and Aβ oligomers. It does not interact with monomer [45,46]. Aducanumab showing positive effect on animal model in improving the cognitive function and decreasing the degree of brain pathology. It can work by stimulation of microglia and prevention of Aβ aggregates evidence of prevention of Aβ by aducanumab is shown in report of study of Tg2576 mice (used as animal model) [2]. In these model Aducanumab reduced the level of Aβ plaques in 9 month old mice in a dose dependent manner but it doesn’t show any significant reduction in Aβ level in 22 month old mice. It means aducanumab prevent the Aβ aggregation more precisely than reduction of Aβ plaque [46]. In 2021, FDA approved aducanumab as the 1st disease modifying therapy for AD. Patient who has Mild Cognitive Impairment (MCI) or who has mild dementia are treated by Aducanumab [2]. The degree of reduction of Aβ in patient and improvement in clinical symptoms is measured by Clinical Dementia Rating Sum of Boxes (CDR-SB) and Mini-Mental State Examination (MMSE) scores. After the successful outcomes obtained in phase 1b of clinical trials, aducunamab goes in phase III in 2015 (clinical trial govt.. A study report of 196 patient shows that aducanumab significantly reduces the Aβ plaques and reduce the clinical measure in patient of AD in same fashion [45,46]. ARIA (Amyloid Related Imaging Abnormalities is the associated side effect seen in dose dependent aducanamab treated group. ARIA occurs generally in those patient who carries gene of Apolipoprotein E [47]. APOE4 is considered as one of the major genetic risk factor in the development of late onset of AD [2].
ARIA is basically comprise of spectrum of imaging that can be detected on MRI. It is developed due to using of monoclonal antibody (aducanumab [48]. There are two types of ARIA may be developed depending on its mechanism [49]. Development of ARIA originates due to antibody mediated breakdown of amyloid plaques and vascular wall structure integrity damage. Amyloid plaques are deposited on the vessel wall and it cause damage of structural integrity, and also reduces the perivascular clearance of amyloid plaques, when monoclonal antibody is administered them breakdown the Aβ and also leads to increase in clearance rate of perivascular drainage. Increase in perivascular drainage leads to more deposition of amyloid plaques on vessel wall, at the same time amyloid mediated inflammation and breakdown of amyloid plaques occurs, these process cause rupture the membrane integrity and cause leakage of proteinaceous fluid and/or RBC into the parenchyma and leptomenginal space. This can cause development of ARIA-E and ARIA-H [50,51].
Classification of ARIA
ARIA-E- Developed due to brain edema or sulcul effusion.
ARIA-H- Developed due to hemorrhage in the brain parenchyma
From June 2021, aducanumab has been approved by FDA to treat mild Alzheimer's disease, which has generated a lot of medical and scientific debate [52]. Even if aducanumab does lower Aβ, there is insufficient solid proof that AD patients actually benefit from treatment. ARIA-related cerebral edema (ARIA-E) occurred in around 35% of patients receiving high- dose aducanumab in phase III, and ARIA-related micro hemorrhages (ARIA-H) or other adverse symptoms, such as headache, nausea, and dizziness, occurred in approximately 18%-22.7% of patients. The majority of ARIA-E episodes happened early on in the treatment with aducanumab. These outcomes were in line with earlier clinical investigations of anti- A antibodies, and subsequent therapy lowered the likelihood of ARIA-E. A review of all trials revealed in a February 2022 Neurology publication that aducanumab dramatically decreased a plaques in the brain. It has not yet been demonstrated, yet, whether it affects symptoms connected to AD. According to reports, brain hemorrhage and swelling occurred in roughly 40% of patients receiving aducanumab treatment, however most side effects vanished once the medication was stopped. Auranumab is currently only licenced for MCI and early-stage AD patients; those with moderate-to-severe AD are not included. FDA advises Magnetic Resonance Imaging (MRI) close monitoring of patients receiving aducanumab, and more thorough research is required on several aspects of aducanumab therapy.
Lecanemab: Lecanemab is newly developed humanized version of the murine mAb158 antibody. Which selectively target the soluble Aβ aggregate (oligomer, and protofibrils) [53]. Soluble Aβ protofibrils are more neurotoxic and it contribute to the pathogenesis of AD [54]. Lecanemab evidences showed that it selectively bind to soluble Aβ aggregate and reduce the pathology of AD, it also prevent the deposition of Aβ and selectively reduce Aβ protofibril in brain and CSF [55,56].
Modest efficacy has been observed in high dose of lecanemab and risk of edema was limited to higher dose of lecanemab in APOE4 carrier [57]. Lecanemab was administered to 609 patients with early-onset AD, MCI, and mild-onset AD dementia, whereas a placebo was given to 245 patients in a randomized double-blind clinical trial, site across North America (the USA and Canada), Europe (France, German, Italy, Netherlands, Spain, Sweden and the UK) and the pacific region (Japan and South Korea) [2,53].
For appropriate subject allocation to the trial's most efficient dose, Bayesian response adaptive randomization was used. The monthly and biweekly 10 mg/kg Early on in the trial, different dosages were identified as potential effective doses of 90% (ED90), with 10 mg/kg administered twice weekly being the final ED90 dose (defined as the most basic treatment group that achieves at least 90% of the modelled maximal treatment impact) [53].
The findings demonstrated that brain Aβ was considerably reduced by lecanemab (10 mg/kg biweekly identified as the target ED90 dose), which was distinct from the placebo group at 72 weeks, supporting the use of lecanemab as an active treatment, but this dose did not received the highest no. of subjects since randomization of ApoE4 carriers [58]. As a results, the lecanemab 10 mg/kg biweekly group had fewer subjects compare to 10 mg/kg monthly with lower percentage of ApoE4 carrier [53].
A Bayesian adaptive design was used in BAN2401-G000-201 (Study 201) with response adaptive randomization, frequent blinded interim analyses to determine early success or futility, and a plan to update subject allocation probabilities based on the anticipated 12-month outcome modelled on all clinical data on the Alzheimer's Disease Composite Score (ADCOMS).
Safety and efficacy of lecanemab are previously performed in clinical trials with mild to moderate AD patients and it was found that lecanemab was well tolerated at all dose [53].
ARIA-E and ARIA-H and infusion reaction was developed but it was well tolerated [54]. The majority of ARIA-E (60%) occurred within the first three months of therapy and had radiologic severity that was primarily mild-to-moderate (89%) in nature. All ARIA-E cases were successfully resolved, typically taking 4 to 12 weeks [53].
Infusion reactions (3.3% for placebo, 5.8% for 2.5 mg/kg biweekly, 7.8% for 5 mg/kg monthly, 12.0% for 5 mg/kg biweekly, 22.9% for 10 mg/kg monthly, and 19.9% for 10 mg/kg biweekly and ARIA-E (0.8% for placebo, 1.9% for 2.5 mg/kg biweekly, 2.0% for 5 mg/kg monthly, 3.3% for 5 mg/kg biweekly, 9.9% for 10 mg/kg monthly, The majority of infusion responses were mild to moderate (Grade 1-2 and frequently improved with preventive therapy. Lecanemab and placebo showed no discernible treatment differences in laboratory tests, electrocardiograms, or vital signs [53].
Changes in CSF biomarkers confirmed the therapeutic benefit of lecanemab, and lecanemab was well-tolerated with a 9.9% incidence of ARIAs-E at 10 mg/kg biweekly [53]. Moreover, lecanemab demonstrated great efficacy in terms of clinical and biomarker outcomes [54]. Lecanemab entirely eliminated A plaques from the brain, slowed cognitive decline, and had a low incidence of ARIA-E in early AD, according to the results of the phases I and II (2b trials [59]. Lecanemab might therefore have an impact on AD pathology to slow down AD development. Lecanemab is a potential feasible mab's medication for the treatment of AD based on encouraging preclinical findings and various clinical trial results. Because of the reduction in brain A levels, improvement in cognitive decline, and low incidence of ARIA-E, lecanemab appears to be the most effective treatment for AD among these mabs. It is safer and has a mild therapeutic effect. The outcomes of numerous clinical trials, however, were generally unfavorable and failed to demonstrate clinically significant improvements in patients with clinically apparent or prodromal dementia. Further research must be done to determine the effectiveness and safety of lecanemab [2].
Binding comparison of aducanumab and lecanemab to different form of Aβ
Using inhibitory ELISA, immunodepletion, and Surface Plasmon Resonance (SPR),the antibodies' binding ability to Aβ monomers, cross-linked oligomers, small and large protofbrils, and fibrils has been studied [42]. Aducanumab selectively binds to fibrils than protofibrils, whereas lecanemab show stronger binding to protofibril than fibrils [60]. By using inhibition ELISA, the binding of lecanemab, aducanumab, and to various in-vitro produced soluble species of Aβ monomers, oligomers, and protofbrils, was studied. For all antibodies, IC50 values were in the µM range for monomeric Aβ. Lecanemab and aducanumab both had IC50 values above>25 µM, which indicated a very weak binding to monomers. Lecanemab had the lowest IC50 values for binding to both small and big protofbrils, measuring 0.8 nM. Aducanumab showed a stronger binding to the large protofbrils with an IC50 of 1.3 nM as compared to the smaller protofbrils with an IC50 of 2.5 Nm [42].
Discussion
In this review we focus on the treatment approach of AD and complications arising during the treatment. Aducanumab is used since 2021 to prevent the accumulation Aβ aggregates plaques more significantly than reducing Aβ aggregates plaques by the FDA approval. ARIA (Amyloid-Related Imaging Abnormalities is the associated side effect seen in dose- dependent Aducanamab patients. Occurrence of ARIA is generally associated with a patients who carries the gene of Apolipoprotein E. Lecanemab is one of the other classes of newly developed humanized versions of the murine mAb158 antibody. It shows to be more effective than Aducanumab because it selectively reduces the soluble Aβ aggregate (oligomer, and protofibrils). Soluble Aβ aggregate is more toxic and it is associated with the generation of the pathogenesis of AD. In the future, we will develop and use the combined form of both the Anti-Aβ Antibodies to successfully treat AD patients.
Conclusion
Alzheimer disease is nowadays biggest emerging disease cause by certain risk factor like diet, genetic factor, infection and environment pollution. The main factor associated for development of AD is accumulation of Aβ plaque in entrorhinal cortex and hippocampus reason. The APP (Amyloid Beta Precursor Protein) under the action of γ-secretase enzyme converted into Aβ. Aβ occurs in many form like soluble aggregate, protofibril, and fibrils. Soluble form is more neurotoxic than other. Tau protein hyperpolarisation is also associated in the development of AD. Monoclonal antibodies like aducanumab, lecaneumab and others are developed in hope for the reducing the AD progression by reducing the Aβ accumulation. Lecanemab, a humanized monoclonal antibody shows effective results in reducing the AD progression.
References
- Tahami AAM, Tafazzoli A, Ye W, Chavan A, Zhang Q. Long-term health outcomes of lecanemab in patients with early alzheimer's disease using simulation modeling. Neurol Ther. 2022;11(2):863-880.
[Crossref] [Google Scholar] [PubMed]
- Shi M, Chu F, Zhu F, Zhu J. Impact of anti-amyloid-β monoclonal antibodies on the pathology and clinical profile of alzheimer's disease: A focus on aducanumab and lecanemab. Front Aging Neurosci. 2022;14:870517.
[Crossref] [Google Scholar] [PubMed]
- Knopman DS, Amieva H, Petersen RC, Chételat G, Holtzman DM, Hyman BT, et al. Alzheimer disease. Nat Rev Dis Primers. 2021 May 13;7(1):33.
[Crossref] [Google Scholar] [PubMed]
- Wu YT, Beiser AS, Breteler MMB, Fratiglioni L, Helmer C, Hendrie HC, et al. The changing prevalence and incidence of dementia over time - current evidence. Nat Rev Neurol. 2017;13(6):327-339.
[Crossref] [Google Scholar] [PubMed]
- Mielke MM, Vemuri P, Rocca WA. Clinical epidemiology of Alzheimer's disease: assessing sex and gender differences. Clin Epidemiol. 2014;6:37-48.
[Crossref] [Google Scholar] [PubMed]
- Thambisetty M, An Y, Tanaka T. Alzheimer's disease risk genes and the age-at-onset phenotype. Neurobiol Aging. 2013;34(11):2696.e1-5.
[Crossref] [Google Scholar] [PubMed]
- Jonsson T, Atwal JK, Steinberg S, Snaedal J, Jonsson PV, Bjornsson S, et al. A mutation in APP protects against Alzheimer's disease and age-related cognitive decline. Nature. 2012;488(7409):96-9.
[Crossref] [Google Scholar] [PubMed]
- Bloom GS. Amyloid-β and tau: the trigger and bullet in Alzheimer disease pathogenesis. JAMA Neurol. 2014 ;71(4):505-8.
[Crossref] [Google Scholar] [PubMed]
- Uddin MS, Al Mamun A, Rahman MA, Behl T, Perveen A, Hafeez A, et al. Emerging Proof of Protein Misfolding and Interactions in Multifactorial Alzheimer's Disease. Curr Top Med Chem. 2020;20(26):2380-2390.
[Crossref] [Google Scholar] [PubMed]
- Kent SA, Spires-Jones TL, Durrant CS. The physiological roles of tau and Aβ: implications for Alzheimer's disease pathology and therapeutics. Acta Neuropathol. 2020;140(4):417-447.
[Crossref] [Google Scholar] [PubMed]
- Mary A, Eysert F, Checler F, Chami M. Mitophagy in Alzheimer's disease: Molecular defects and therapeutic approaches. Mol Psychiatry. 2023 Jan;28(1):202-216.
[Crossref] [Google Scholar] [PubMed]
- Amadoro G, Corsetti V, Florenzano F, Atlante A, Ciotti MT, Mongiardi MP, et al. AD-linked, toxic NH2 human tau affects the quality control of mitochondria in neurons. Neurobiol Dis. 2014;62:489-507.
[Crossref] [Google Scholar] [PubMed]
- Khan UA, Liu L, Provenzano FA, Berman DE, Profaci CP, Sloan R, et al. Molecular drivers and cortical spread of lateral entorhinal cortex dysfunction in preclinical Alzheimer's disease. Nat Neurosci. 2014;17(2):304-11.
[Crossref] [Google Scholar] [PubMed]
- Lu JX, Qiang W, Yau WM, Schwieters CD, Meredith SC, Tycko R. Molecular structure of β-amyloid fibrils in Alzheimer's disease brain tissue. Cell. 2013;154(6):1257-68.
[Crossref] [Google Scholar] [PubMed]
- O'Nuallain B, Freir DB, Nicoll AJ, Risse E, Ferguson N, Herron CE, et al. Amyloid beta-protein dimers rapidly form stable synaptotoxic protofibrils. J Neurosci. 2010;30(43):14411-9.
[Crossref] [Google Scholar] [PubMed]
- Paravastu AK, Leapman RD, Yau WM, Tycko R. Molecular structural basis for polymorphism in Alzheimer's beta-amyloid fibrils. Proc Natl Acad Sci U S A. 2008;105(47):18349-54.
[Crossref] [Google Scholar] [PubMed]
- Yu X, Zheng J. Polymorphic structures of Alzheimer's β-amyloid globulomers. PLoS One. 2011;6(6):e20575.
[Crossref] [Google Scholar] [PubMed]
- Haass C, Willem M. Secreted APP Modulates Synaptic Activity: A Novel Target for Therapeutic Intervention? Neuron. 2019 Feb 20;101(4):557-559.
[Crossref] [Google Scholar] [PubMed]
- Huynh TV, Davis AA, Ulrich JD, Holtzman DM. Apolipoprotein E and Alzheimer's disease: the influence of apolipoprotein E on amyloid-β and other amyloidogenic proteins. J Lipid Res. 2017;58(5):824-836.
[Crossref] [Google Scholar] [PubMed]
- Kim J, Basak JM, Holtzman DM. The role of apolipoprotein E in Alzheimer's disease. Neuron. 2009;63(3):287-303.
[Crossref] [Google Scholar] [PubMed]
- Zhou R, Ji B, Kong Y, Qin L, Ren W, Guan Y, et al. PET Imaging of Neuroinflammation in Alzheimer's Disease. Front Immunol. 2021;12:739130.
[Crossref] [Google Scholar] [PubMed]
- Albert MS, DeKosky ST, Dickson D, Dubois B, Feldman HH, Fox NC, et al. The diagnosis of mild cognitive impairment due to Alzheimer's disease: recommendations from the National Institute on Aging-Alzheimer's Association workgroups on diagnostic guidelines for Alzheimer's disease. Alzheimers Dement. 2011;7(3):270-9.
[Crossref] [Google Scholar] [PubMed]
- Buée L, Bussière T, Buée-Scherrer V, Delacourte A, Hof PR. Tau protein isoforms, phosphorylation and role in neurodegenerative disorders. Brain Res Brain Res Rev. 2000;33(1):95-130.
[Crossref] [Google Scholar] [PubMed]
- Malpetti M, Kievit RA, Passamonti L, Jones PS, Tsvetanov KA, Rittman T, et al. Microglial activation and tau burden predict cognitive decline in Alzheimer's disease. Brain. 2020;143(5):1588-1602.
[Crossref] [Google Scholar] [PubMed]
- Parbo P, Ismail R, Hansen KV, Amidi A, Mårup FH, Gottrup H, et alBrain inflammation accompanies amyloid in the majority of mild cognitive impairment cases due to Alzheimer's disease. Brain. 2017;140(7):2002-2011.
[Crossref] [Google Scholar] [PubMed]
- Hagens MHJ, Golla SV, Wijburg MT, Yaqub M, Heijtel D, Steenwijk MD, et al. In vivo assessment of neuroinflammation in progressive multiple sclerosis: a proof of concept study with [18F]DPA714 PET. J Neuroinflammation. 2018;15(1):314.
[Crossref] [Google Scholar] [PubMed]
- Hamelin L, Lagarde J, Dorothée G, Potier MC, Corlier F, Kuhnast B, et al. Distinct dynamic profiles of microglial activation are associated with progression of Alzheimer's disease. Brain. 2018;141(6):1855-1870.
[Crossref] [Google Scholar] [PubMed]
- Qiao L, Fisher E, McMurray L, Milicevic Sephton S, Hird M, Kuzhuppilly-Ramakrishnan N, et al. Radiosynthesis of (R,S)-[18 F]Ge387: a potential pet radiotracer for imaging translocator protein 18 KDA (TSPO) with low binding sensitivity to the human gene polymorphism rs6971. ChemMedChem. 2019;14(9):982-993.
[Crossref] [Google Scholar] [PubMed]
- Butsch V, Börgel F, Galla F, Schwegmann K, Hermann S, Schäfers M, et al. Design, (radio)synthesis, and in vitro and in vivo evaluation of highly selective and potent matrix metalloproteinase 12 (MMP-12) inhibitors as radiotracers for positron emission tomography. J Med Chem. 2018;61(9):4115-4134.
[Crossref] [Google Scholar] [PubMed]
- Kasten BB, Jiang K, Cole D, Jani A, Udayakumar N, Gillespie GY, et al. Targeting MMP-14 for dual PET and fluorescence imaging of glioma in preclinical models. Eur J Nucl Med Mol Imaging. 2020;47(6):1412-1426.
[Crossref] [Google Scholar] [PubMed]
- Zheng QH, Fei X, DeGrado TR, Wang JQ, Stone KL, Martinez TD, et al. Synthesis, biodistribution and micro-PET imaging of a potential cancer biomarker carbon-11 labeled MMP inhibitor (2R)-2-[[4-(6-fluorohex-1-ynyl)phenyl]sulfonylamino]-3-methylbutyric acid [11C]methyl ester. Nucl Med Biol. 2003;30(7):753-60.
[Crossref] [Google Scholar] [PubMed]
- Luzi F, Savickas V, Taddei C, Hader S, Singh N, Gee AD, et al. Radiolabeling of [11C] FPS-ZM1, a receptor for advanced glycation end products-targeting positron emission tomography radiotracer, using a [11C] CO2-to-[11C] CO chemical conversion. Future Med. 2020;12(6):511-21.
- Olivares D, Deshpande VK, Shi Y, Lahiri DK, Greig NH, Rogers JT, et al. N-methyl D-aspartate (NMDA) receptor antagonists and memantine treatment for Alzheimer's disease, vascular dementia and Parkinson's disease. Curr Alzheimer Res. 2012;9(6):746-58.
[Crossref] [Google Scholar] [PubMed]
- Arvanitakis Z, Shah RC, Bennett DA. Diagnosis and management of dementia: Review. JAMA. 2019;322(16):1589-1599.
[Crossref] [Google Scholar] [PubMed]
- Cummings JL, Tong G, Ballard C. Treatment Combinations for Alzheimer's Disease: Current and Future Pharmacotherapy Options. J Alzheimers Dis. 2019;67(3):779-794.
[Crossref] [Google Scholar] [PubMed]
- Hartley DM, Walsh DM, Ye CP, Diehl T, Vasquez S, Vassilev PM, et al. Protofibrillar intermediates of amyloid beta-protein induce acute electrophysiological changes and progressive neurotoxicity in cortical neurons. J Neurosci. 1999;19(20):8876-84.
[Crossref] [Google Scholar] [PubMed]
- Paranjape GS, Gouwens LK, Osborn DC, Nichols MR. Isolated amyloid-β(1-42) protofibrils, but not isolated fibrils, are robust stimulators of microglia. ACS Chem Neurosci. 2012;3(4):302-11.
[Crossref] [Google Scholar] [PubMed]
- Sehlin D, Englund H, Simu B, Karlsson M, Ingelsson M, Nikolajeff F, et al. Large aggregates are the major soluble Aβ species in AD brain fractionated with density gradient ultracentrifugation. PLoS One. 2012;7(2):e32014.
[Crossref] [Google Scholar] [PubMed]
- Zhu F, Li C, Chu F, Tian X, Zhu J. Target Dysbiosis of Gut Microbes as a Future Therapeutic Manipulation in Alzheimer's Disease. Front Aging Neurosci. 2020;12:544235.
[Crossref] [Google Scholar] [PubMed]
- Guillot-Sestier MV, Town T. Innate immunity in Alzheimer's disease: A complex affair. CNS Neurol Disord Drug Targets. 2013;12(5):593-607.
[Crossref] [Google Scholar] [PubMed]
- Webers A, Heneka MT, Gleeson PA. The role of innate immune responses and neuroinflammation in amyloid accumulation and progression of Alzheimer's disease. Immunol Cell Biol. 2020;98(1):28-41.
[Crossref] [Google Scholar] [PubMed]
- Söderberg L, Johannesson M, Nygren P, Laudon H, Eriksson F, Osswald G, et al. Lecanemab, Aducanumab, and Gantenerumab - Binding profiles to different forms of amyloid-beta might explain efficacy and side effects in clinical trials for alzheimer's disease. Neurotherapeutics. 2023;20(1):195-206.
[Crossref] [Google Scholar] [PubMed]
- Miles LA, Crespi GA, Doughty L, Parker MW. Bapineuzumab captures the N-terminus of the Alzheimer's disease amyloid-beta peptide in a helical conformation. Sci Rep. 2013;3:1302.
[Crossref] [Google Scholar] [PubMed]
- Honig LS, Vellas B, Woodward M, Boada M, Bullock R, Borrie M, et al. Trial of Solanezumab for Mild Dementia Due to Alzheimer's Disease. N Engl J Med. 2018;378(4):321-330.
[Crossref] [Google Scholar] [PubMed]
- Linse S, Scheidt T, Bernfur K, Vendruscolo M, Dobson CM, Cohen SIA, et al. Kinetic fingerprints differentiate the mechanisms of action of anti-Aβ antibodies. Nat Struct Mol Biol. 2020;27(12):1125-1133.
[Crossref] [Google Scholar] [PubMed]
- Sevigny J, Chiao P, Bussière T, Weinreb PH, Williams L, Maier M, et al The antibody aducanumab reduces Aβ plaques in Alzheimer's disease. Nature. 2016;537(7618):50-6.
[Crossref] [Google Scholar] [PubMed]
- Cummings JL, Cohen S, van Dyck CH, Brody M, Curtis C, Cho W et al. ABBY: A phase 2 randomized trial of crenezumab in mild to moderate Alzheimer disease. Neurology. 2018;90(21):e1889-e1897.
[Crossref] [Google Scholar] [PubMed]
- Gaillard F, Sharma R. Amyloid related imaging abnormalities (ARIA). Radiopaedia.Org.2023.
[Crossref]
- Nisticò R, Borg JJ. Aducanumab for Alzheimer's disease: A regulatory perspective. Pharmacol Res. 2021 Sep;171:105754.
[Crossref] [Google Scholar] [PubMed]
- Barakos J, Purcell D, Suhy J, Chalkias S, Burkett P, Marsica Grassi C, et al. Detection and management of amyloid-related imaging abnormalities in patients with Alzheimer’s disease treated with anti-amyloid beta therapy. J Prev Alzheimers Dis. 2022;9(2):211-220.
[Crossref] [Google Scholar] [PubMed]
- Sperling R, Salloway S, Brooks DJ, Tampieri D, Barakos J, et al. Amyloid-related imaging abnormalities in patients with Alzheimer's disease treated with Bapineuzumab: A retrospective analysis. Lancet Neurol. 2012;11(3):241-9.
[Crossref] [Google Scholar] [PubMed]
- Dhillon S. Aducanumab: First Approval. Drugs. 2021;81(12):1437-1443.
[Crossref] [Google Scholar] [PubMed]
- Swanson CJ, Zhang Y, Dhadda S, Wang J, Kaplow J, Lai RYK, et al . A randomized, double-blind, phase 2b proof-of-concept clinical trial in early Alzheimer's disease with lecanemab, an anti-Aβ protofibril antibody. Alzheimers Res Ther. 2021;13(1):80.
[Crossref] [Google Scholar] [PubMed]
- Logovinsky V, Satlin A, Lai R, Swanson C, Kaplow J, Osswald G, et al. Safety and tolerability of BAN2401--a clinical study in Alzheimer's disease with a protofibril selective Aβ antibody. Alzheimers Res Ther. 2016;8(1):14.
[Crossref] [Google Scholar] [PubMed]
- Söllvander S, Ekholm-Pettersson F, Brundin RM, Westman G, Kilander L, Paulie S, et al. Increased Number of Plasma B Cells Producing Autoantibodies Against Aβ42 Protofibrils in Alzheimer's Disease. J Alzheimers Dis. 2015;48(1):63-72.
[Crossref] [Google Scholar] [PubMed]
- Tucker S, Möller C, Tegerstedt K, Lord A, Laudon H, Sjödahl J, et al. The murine version of BAN2401 (mAb158) selectively reduces amyloid-β protofibrils in brain and cerebrospinal fluid of tg-ArcSwe mice. J Alzheimers Dis. 2015;43(2):575-88.
[Crossref] [Google Scholar] [PubMed]
- Abushakra S, Porsteinsson A, Vellas B, Cummings J, Gauthier S, Hey JA, et al. Clinical Benefits of Tramiprosate in Alzheimer's Disease Are Associated with Higher Number of APOE4 Alleles: The "APOE4 Gene-Dose Effect". J Prev Alzheimers Dis. 2016;3(4):219-228.
[Crossref] [Google Scholar] [PubMed]
- Swanson CJ, Zhang Y, Dhadda S, Wang J, Kaplow J, Lai RYK, et al. A randomized, double-blind, phase 2b proof-of-concept clinical trial in early Alzheimer's disease with lecanemab, an anti-Aβ protofibril antibody. Alzheimers Res Ther. 2022;14(1):70. doi: 10.1186/s13195-022-00995-9.
[Crossref] [Google Scholar] [PubMed]
- Wang J, Logovinsky V, Hendrix SB, Stanworth SH, Perdomo C, Xu L, et al. ADCOMS: A composite clinical outcome for prodromal Alzheimer's disease trials. J Neurol Neurosurg Psychiatry. 2016;87(9):993-9.
[Crossref] [Google Scholar] [PubMed]
- Magnusson K, Sehlin D, Syvänen S, Svedberg MM, Philipson O, Söderberg L, et al. Specific uptake of an amyloid-β protofibril-binding antibody-tracer in AβPP transgenic mouse brain. J Alzheimers Dis. 2013;37(1):29-40.
[Crossref] [Google Scholar] [PubMed]
Citation: Verma A, Rosi, Sharma P (2023) A Systemic Review on Aducanumab and Lecanamab in the Development of ARIA in Alzheimer’s disease. J Clin Exp Pharmacol. 13:393.
Copyright: © 2023 Verma A, et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.